

1 Introduction
The number of tailor-made transition-metal catalysts for various chemical transformations is ever increasing [1], but the practical use of these homogenous catalysts is occasionally hampered by the cumbersome separation of the catalyst from the product-phase [2]. Various new procedures for catalyst recycling have been investigated [3, 4], supported aqueous phase catalysis [5], fluorous phase catalysis [6, 7], the use of ionic liquids [8] and supercritical fluids [9, 10]. A widely studied approach to facilitate catalyst-product separation is the attachment of homogeneous catalysts to dendritic [11–16], or hybrid supports [17–20]. We have studied several approaches to obtain recyclable catalyst systems, including immobilization on various supports and two-phase catalysis [21–30]. As an important alternative we have been investigating the immobilization of catalysts to dendritic support. In principle, dendritic catalysts should provide systems that show the kinetic behaviour and thus the activity and selectivity of a conventional homogeneous catalyst, but additionally can be removed easily from the reaction mixture using nanofiltration techniques. In addition, an interesting unanswered question when we started this project was if a dendritic catalyst would show catalytic behaviour different from the homogeneous parent metal-complex. If we would understand these dendritic effects and would be able to use the dendritic effects to our advantage, we would have created a new tool to control important catalytic properties such as selectivity, activity and stability. Since these novel properties, induced by the dendritic framework, obviously depend on the location of the functional group within the structure, we prepared both periphery- (Fig. 1, 1–16) and core-functionalized dendritic catalysts (Figs. 2–4, 17–30) based on carbosilane dendrimers. In this review we will summarize our most important results.
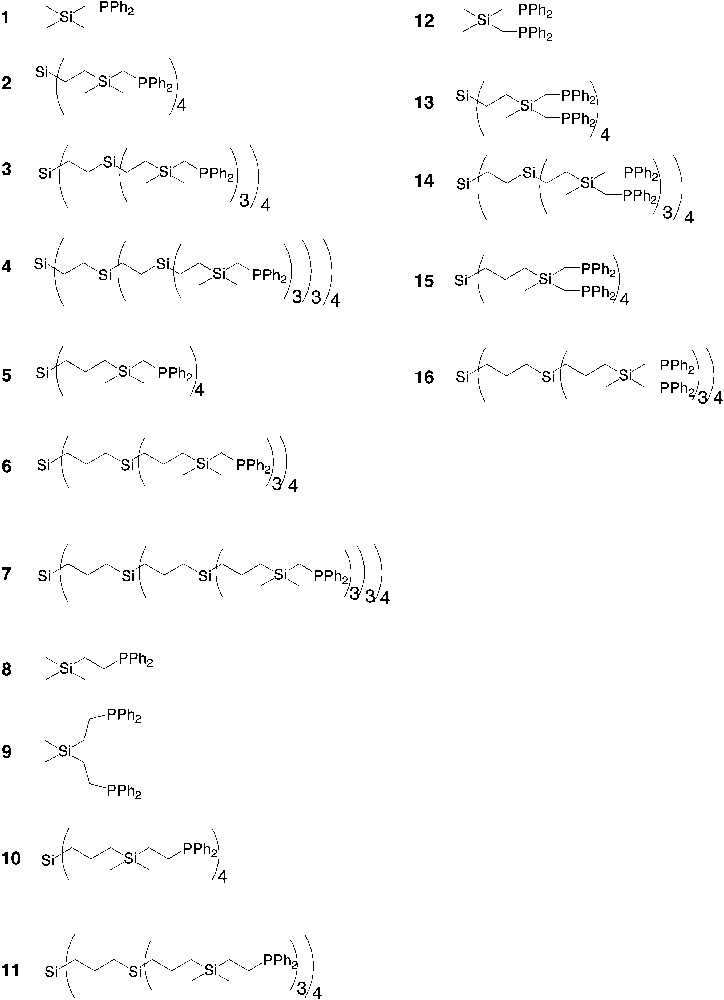
Diphenylphosphine-functionalized dendrimers that have been used as multi-dentate ligands in catalysis. The number of phosphine per end-group is one (1–11) or two (12–16).

Carbosilane dendrimers with dppf-type ligands located in the core.
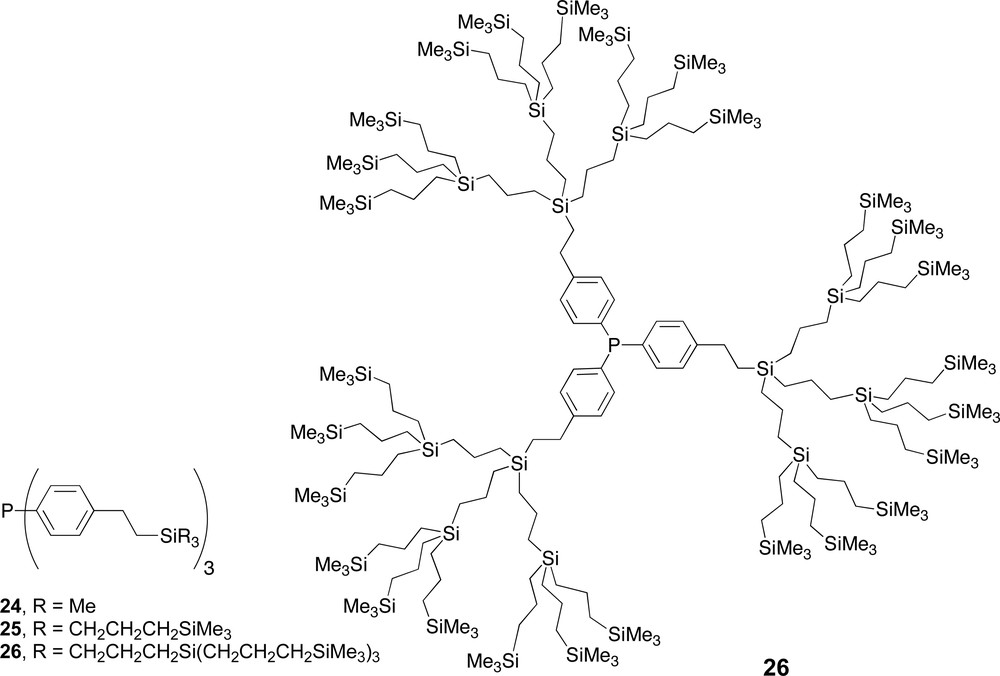
Carbosilane dendrimers with phosphine ligands located in the core.
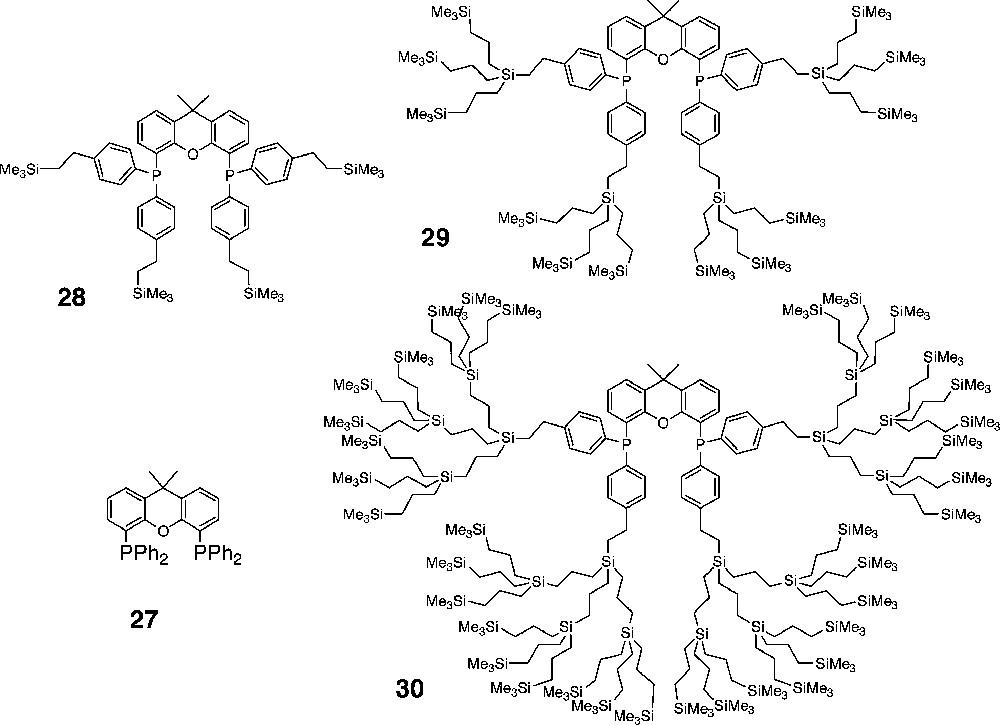
Carbosilane dendrimers with xantphos (27)-type ligands located in the core.
2 Rhodium catalyzed hydroformylation with dendritic phosphine catalyst
The hydroformylation [31] reaction is an important chemical transformation in which an alkene is converted into an aldehyde (Fig. 5). Important issues concern the activity of the catalyst and the control of the regioselectivity of the reaction. Depending on the ligands, rhodium-based complexes can give fast and selective catalysts under mild conditions. Diphosphine ligands having large natural bite angles (P–Rh–P) [32], e.g., xantphos-type ligands, give very high selectivities to the linear product in the hydroformylation of 1-octene [33–34]. The number of examples of dendrimer-supported catalysts that have been used for the hydroformylation reaction is surprisingly limited [35–40, 54, 55]. We have studied both periphery- and core- functionalized dendrimers as catalysts for this reaction.
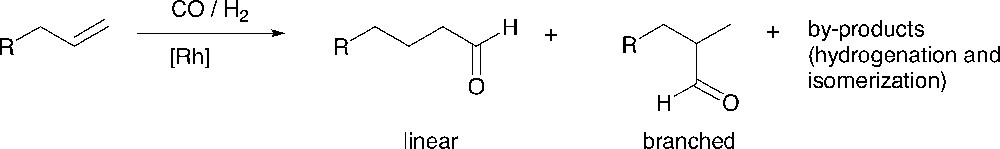
The hydroformylation reaction.
2.1 Periphery functionalized dendrimers
Diphenylphosphine-terminated carbosilane dendrimers were used as ligands in the rhodium catalyzed hydroformylation of 1-octene and the results are summarized in Table 1a [41, 42]. In general, we observe that higher phosphine/rhodium ratios result in lower activities for the dendritic ligands. This is in contrast with the monomeric model compound (CH3)3SiCH2PPh2 (1), which gives a more active catalyst at a higher P/Rh ratio (Table 1a). This is an effect of the high local phosphine concentration of the dendritic ligands, which results in less active rhodium complexes with three or four coordinating phosphine ligands. Furthermore, high local concentration of rhodium might lead to carbonyl-bridged dimeric rhodium complexes, which generally show little or no activity.
Rhodium catalyzed hydroformylation of 1–octene using various dendritic ligandsa
(a) Dendrimers with Si(CH3)2CH2PPh2 end-groups
| Ligand | P/Rh | Time (h) | Conversion (%) | l/b | |
| 1 | 10 | 0.5 | 82 | 2.3 | |
| 5 | 0.5 | 63 | 2.3 | ||
| 2.5 | 0.5 | 40 | 2.3 | ||
| 2.5 | 1 | 97 | 2.3 | ||
| 2 | 10 | 1 | 24 | 2.3 | |
| 5 | 1 | 56 | 2.3 | ||
| 2.5 | 1 | 69 | 2.3 | ||
| 3 | 10 | 1 | 13 | 2.4 | |
| 5 | 1 | 17 | 2.4 | ||
| 2.5 | 1 | 24 | 2.4 | ||
| 4 | 10 | 1 | 12 | 2.4 | |
| 5 | 1 | 14 | 2.4 | ||
| 2.5 | 1 | 24 | 2.4 | ||
| 5 | 10 | 1 | 40 | 2.4 | |
| 5 | 1 | 55 | 2.3 | ||
| 2.5 | 1 | 64 | 2.3 | ||
| 6 | 10 | 1 | 22 | 2.4 | |
| 5 | 1 | 23 | 2.4 | ||
| 2.5 | 1 | 59 | 2.3 | ||
| 7 | 10 | 1 | 18 | 2.4 | |
| 5 | 1 | 23 | 2.4 | ||
| 2.5 | 1 | 41 | 2.3 |
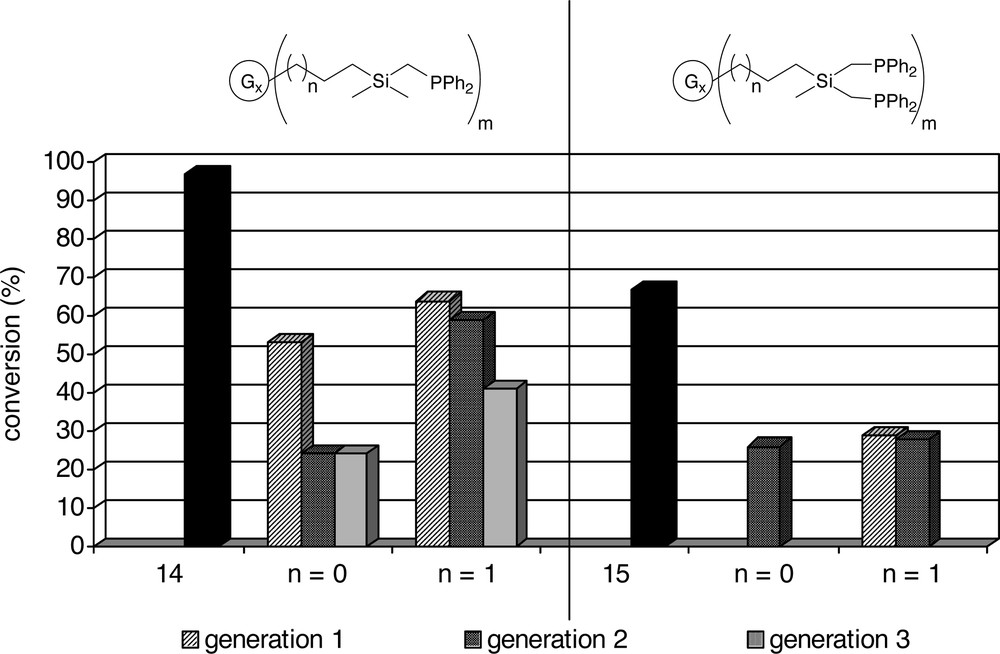
Dendrimers with –(CH3)2SiCH2PPh2 end-groups (Table 1a) give generally faster catalysts than the dendrimers with –(CH3)Si(CH2PPh2)2 end-groups (Table 1b), a difference that is also observed for the model compounds (1 and 12). For P/Rh ratios of 5 and 2.5 the catalysts containing the dendritic ligands with –(CH3)Si(CH2PPh2)2 end-groups give very similar conversions (Table 1b), all being about three times as low as the conversion of the model compound (12). The high isomerisation observed for ligand 13 indicates the presence of phosphine free rhodium. For a P/Rh ratio of 10 there is a slight decrease in activity upon going from the first generation to the second-generation dendrimers (14 < 13 and 16 < 15), while the difference between the first generation dendrimers (13 and 15) and the model compound (12) is negligible.
Rhodium catalyzed hydroformylation of 1–octene using various dendritic ligandsa
(b) Dendrimers with Si(CH3)(CH2PPh2)2 end-groups
| Ligand | P/Rh | Time (h) | Conversion (%) | l/b | |
| 12 | 10 | 4 | 16 | 2.4 | |
| 5 | 4 | 92 | 2.6 | ||
| 2.5 | 1 | 67 | 2.4 | ||
| 13 | 10 | 4 | 14 | 2.2 | |
| 5 | 4 | 22 | 2.3 | ||
| 2.5 | 1 | n.d.b | |||
| 14 | 10 | 4 | 7 | 2.2 | |
| 5 | 4 | 41 | 2.3 | ||
| 2.5 | 1 | 26 | 2.4 | ||
| 15 | 10 | 4 | 13 | 2.1 | |
| 5 | 4 | 34 | 2.2 | ||
| 2.5 | 1 | 29 | 2.4 | ||
| 16 | 10 | 4 | 6 | 1.9 | |
| 5 | 4 | 31 | 2.6 | ||
| 2.5 | 1 | 28 | 2.4 |
Variation of the dendritic backbone has a slight influence on the catalytic performance: for the systems functionalized with –(CH3)2SiCH2PPh2 end-groups (Table 1a), the C3-dendrimers (5–7) give faster catalysts than the more compact C2-dendrimers (2–4) (see also Fig. 6). This effect is less pronounced for the first generation dendrimers (2 and 5). For a P/Rh ratio of 5 and 2.5, the activities of these systems are similar. However, when a P/Rh 10 is used, C3-dendrimer (5), in which the phosphines are further apart than in the more compact C2-dendrimer, gives a more active catalyst than 2 (40% conversion versus 24%). For the second and third generation dendrimers, where the local concentration of phosphines is higher, the more compact C2-dendrimers (3 and 4) give slower catalysts than the more flexible C3-dendrimers (6 and 7).
Conversion of 1-octene in the Rh-catalyzed hydroformylation of 1-octene (P/Rh = 2.5) using various carbosilane dendrimers.
Alper and co-workers performed hydroformylation reactions using rhodium complexes of N(CH2PPh2)2-functionalized poly(amido amine) dendrimers on silica. They reported an increase in activity for dendrimers with a larger chain length [36, 38]. The similarity of the results using the –(CH3)Si(CH2PPh2)2-terminated dendrimers (13, 14, 15 and 16) indicates that in the homogeneous phase we do not observe such a difference. The variation in activity for the dendritic catalysts with –(CH3)2SiCH2PPh2 end-groups is likely to be due to the change in distance between the individual phosphines and thus the ring size of the (dendrimer)P–Rh–P ring. In the compact C2-dendrimers the phosphines are forced in even closer proximity to one another than in the C3-dendrimers, which is reflected in a generally lower activity for the former.
Table 2 shows the hydroformylation results using the dendrimers with –Me2SiCH2CH2PPh2 end-groups as the ligands. The SiCH2CH2PPh2-model compound (8) give very similar results to the SiCH2PPh2-model compound (1), as can be expected from monodentate ligands. The dendritic ligands (10 and 11), however, give much slower catalysts than their SiCH2PPh2-analogues (5 and 6). Interestingly, the dendritic ligand with twelve SiCH2CH2PPh2 end-groups (11) induces a higher selectivity for the linear aldehyde (l/b = 4.9). This is a dendritic effect similar to that observed by Cole-Hamilton and co-workers for dendritic systems containing Si(CH3)(CH2CH2PPh2)2 end-groups. They found a linear to branched ratio of 13.9 for a silsesquioxane-cored carbosilane dendrimer with 16 phosphine end-groups [40]. When similar reaction conditions were applied to our systems (10 bar H2/CO, 120 °C, P/Rh ratio of 6, Rh-complex of 11) we obtained a slightly higher l/b compared to our standard conditions (l/b = 5.3 at 60% conversion after 1 h), but also the amount of isomerised 1-octene(educt) increased.
Rhodium-catalyzed hydroformylation of 1-octene using dendritic ligands with an ethylene-spacer between the phosphine and silicon end-groupsa
| Ligand | P/Rh | Time (h) | Conversion (%) | Linear aldehyde (%) | Branched aldehyde (%) | l/b |
| 8 | 2.5 | 1 | 98 | 70 | 30 | 2.3 |
| 9 | 2.5 | 1 | 62 | 72 | 28 | 2.6 |
| 10 | 2.5 | 1 | 16 | 74 | 26 | 2.8 |
| 11 | 2.5 | 1 | 11 | 83 | 17 | 4.9 |
Although the effects in the hydroformylation are not huge, it is interesting to observe dendritic effects in catalysis using periphery-functionalized systems. We observed in some instances lower activities due to high local concentrations of ligands, but also an increase in the selectivity of the reaction can be ascribed to the effect of the dendrimer. Detailed studies are required to exactly understand the effects.
2.2 Core-functionalized dendrimers
The results of the hydroformylation of 1-octene using the three series of dendritic ligands and the corresponding parent ligands are summarized in Table 3 [43]. All dendritic ligands gave active hydroformylation catalysts. No hydrogenation side-products and only small amounts of isomerisation of the substrate to internal alkenes were observed.
The hydroformylation of 1–octene using the dendritic ligands based on triphenylphosphine, xantphos, and dppf.a
| Entry | Ligand | TOFb (× 102) | l/b ratioc | Linear aldehyde (%)d | Isomerisation (%)e |
| 1 | PPh3f | 29 | 2.9 | 73 | 2.3 |
| 2 | 24f | 28 | 2.2 | 69 | 2.6 |
| 3 | 25f | 27 | 2.6 | 71 | 2.6 |
| 4 | 26f | 16 | 2.6 | 71 | 1.9 |
| 5 | 27g | 1.7 | 53 | 95 | 3.8 |
| 6 | 28g | 1.8 | 56 | 95 | 3.9 |
| 7 | 29g | 1.3 | 56 | 95 | 3.7 |
| 8 | 30g | 1.7 | 53 | 94 | 3.9 |
| 9 | 17 (dppf)h | 8.2 | 3.8 | 76 | 3.8 |
| 10 | Dtpfh,i | 7.7 | 3.7 | 75 | 4.1 |
| 11 | 18bh | 7.8 | 2.6 | 66 | 6.5 |
| 12 | 19bh | 5.4 | 3.7 | 76 | 3.9 |
| 13 | 20h | 4.5 | 3.2 | 72 | 5.9 |
Application of the dendritic monophosphine ligands 24 and 25 resulted in similar activities compared to triphenylphosphine, whereas for the largest dendrimer 26 the activity decreased with a factor of two (entries 1–4). The observed decrease in reaction rate when 26 was applied can be ascribed to the dendritic encapsulation of the catalytic centre, which hinders the approach of the substrate. Interestingly, the selectivity and the amount of isomerisation remain unchanged compared to triphenylphosphine. This indicates that under the reaction conditions still two phosphine ligands 26 are coordinated to the rhodium centre, giving a complex with a molecular mass around 8500 D. All the monophosphine ligands 24–26 induce the expected regioselectivities for the linear aldehyde similar to PPh3.
The ligands based on the rigid xanthene backbone all show a high preference for the formation of the linear aldehyde (95%). A high linear to branched (l/b) ratio of around 55 is found for all generations dendritic ligands (entries 5–8, Table 3), which is similar to that induced by the parent xantphos ligand [33,34]. This high selectivity is a result of the large P–Rh–P bite angle (112°), which determines the steric properties around the catalytic centre. The regioselectivity in the hydroformylation of 1-octene is determined by the rigid ligand in the core and is not affected by the size of the dendritic encapsulation. Moreover, the activity of the catalyst is hardly affected by the dendrimer. The catalytic performance using the rhodium complex based on 30 is very similar to that of the complex of the parent ligand, but interestingly the dimensions of the dendrimer-supported catalysts are in the range that efficient separation by nanofiltration becomes feasible.
Rhodium complexes of the dendritic ligands based on the more flexible dppf-core were studied in the hydroformylation of 1-octene and compared with dtpf (1,1′-bis[di-p-tolylphosphino]ferrocene) to evaluate the effect of para-substitution (Table 3, entries 9–13). The para-substitution slightly affected the activity of the catalyst, which is in line with that previously found by others [44]. Generally the activities of these systems were in between those of the triphenylphosphine and the xantphos-based catalysts. Moreover, we found a decrease in reaction rate of a factor of two compared to dppf when the largest ferrocenyl dendrimer 20 was applied, an effect that is similar to that of the dendritic monophosphines. The dppf-type ligands induced a selectivity of 66–76% for the linear aldehyde and circa 4–6% isomerised alkene was obtained. Generally the ferrocenyl diphosphine dendrimers induce the same selectivity as the non-encapsulated ligands dppf and dtpf. Interestingly, in the hydroformylation of sterically demanding substrates such as 4,4,4-triphenylbut-1-ene the activity drops with a factor of 4 when higher generation dendritic ligands are used, an effect that is larger than for the smaller substrate 1-octene. Potentially this difference can be used for substrate discrimination based on size.
The experiments using the different core functionalized dendrimers in the hydroformylation reaction with different substrates show that the selectivity and activity is largely determined by the ligand backbone. The steric bulk of the dendrimer around the catalyst can result in a decrease in the rate of the hydroformylation, but the extent clearly depends on the exact structure of the ligand system. This is important regarding the optimum size of the dendritic catalyst with respect to activity and recyclability of the system.
3 Allylic alkylation
Palladium-catalyzed C–C bond formation reactions are powerful tools in organic synthesis, providing mild and selective methods for the production of a great variety of valuable chemicals from basic precursors. An intriguing example of such a reaction is the allylic substitution reaction pioneered by Trost and others, which led to catalyst systems that have been extensively used in modern organic synthesis [45–47].
We studied the application of both periphery- [48, 49] and core-functionalized dendritic systems [50] in the allylic substitution using continuous and batch processes. The stability of the catalyst appeared to be an important issue, especially with respect to practical applications of these types of immobilized systems (Table 4).
Pd-catalyzed allylic alkylation of crotyl acetate or cinnamyl acetate and sodium diethyl 2-methylmalonate, using Pd(allyl)-complexes of 2, 4, 13, 14
| Substrate | Ligand | Conversion (%) | trans (%) | cis (%) | Branched(%) |
| crotyl acetatea | 2 | 2 | 80 | 5 | 15 |
| (R=Me) | 3 | 2 | 78 | 6 | 16 |
| 13 | 30 | 80 | 5 | 15 | |
| 14 | 22 | 80 | 5 | 15 | |
| cinnamyl acetateb | 2 | 0.6 | 99 | 0 | 1 |
| (R=Ph) | 3 | 0.6 | 100 | 0 | 0 |
| 13 | 10 | 97 | 0 | 3 | |
| 14 | 10 | 96 | 0 | 4 |
3.1 Periphery-functionalized dendrimers
3.1.1 Batch process
The Pd(allyl) complexes of dendrimers 2, 4, 13 and 14 were used as catalysts in the allylic alkylation reaction of substituted allyl acetates with sodium diethyl 2-methylmalonate as the nucleophile (Fig. 7).
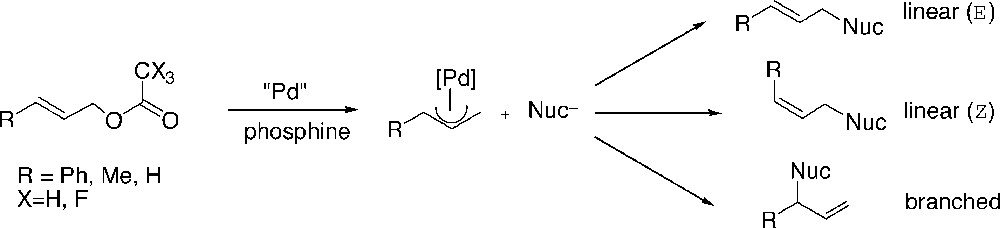
Pd-catalyzed allylic alkylation.
The catalysts having dendritic ligands with a large bridge between the P-atoms (2 and 4) have a very low activity compared to the catalysts containing 13 and 14 as ligands (Table 4). The selectivity towards the linear–trans product is the same for all catalysts and is also similar to that induced by palladium complexes of dppp (= bis(diphenylphosphino)propane) or dppb (= bis(diphenylphosphino)butane) [51]. The second-generation dendrimer 14 gives a less active catalyst for the reaction with crotyl acetate than its smaller analogue 13. For cinnamyl acetate these dendrimers give catalysts with similar activity.
Allyl trifluoroacetate was used as a substrate in a continuous-flow membrane reactor since this leads to the formation of soluble sodium trifluoroacetate as side-product. We studied this substrate also in a batch process and all the dendritic catalysts showed a very high activity. Using a substrate-Pd ratio of 2000 the yield after 5 min was approximately 50% and only small differences in reaction rates were observed for the different generations; the conversions after 5 min were 49%, 55%, 45% and 47% using ligands 2, 4, 13 and 14, respectively. The activity did not decrease with increasing generation, which indicates that all active sites act as independent catalysts.
Addition of a second portion of substrate after a conversion of over 90% showed that the catalyst remained active. Turnover numbers of over 15 000 were reached in the batch process using 14-(Pd[allyl]Cl)12, suggesting that this dendritic catalyst is sufficiently stable for application in a continuous process using a membrane reactor.
3.1.2 Application in a continuous-flow membrane reactor
For the continuous process, a solution of allyl trifluoroacetate and sodium diethyl 2-methylmalonate in THF (including n-decane as an internal standard) was pumped through the reactor containing 14-(Pd[allyl]Cl)12 as the catalyst.
Fig. 8 shows the yield as function of the amount of substrate solution (expressed in reactor volumes) pumped through the reactor. The reaction started immediately after addition of the catalyst and reached its maximum yield after one reactor volume. The yield slowly dropped to zero after approximately 15 times the reactor volume of substrate solution had been pumped through the reactor. Regarding the size of the system and the stability of the catalyst in batch reactions, this decrease was unexpectedly rapid. Based on the retention (99.7% in dichloromethane), the expected decrease in yield was less than 5%. The observed decrease in catalyst activity is therefore ascribed to decomposition of the palladium compound and not to loss of the dendritic catalyst. This is in agreement with the observation that samples taken from the product flow were not catalytically active, indicating that no active palladium catalyst had passed through the membrane. When the experiment was stopped after 39 reactor volumes of substrate solution had been flushed through the reactor, the contents of the reactor and the solution that had been passed through the membrane were analysed for palladium. ICP–AES showed that all the palladium had passed through the membrane. Batch experiments performed in the presence of pieces of membrane material gave similar results as those without membrane suggesting that the membrane material did not play a role in the deactivation process. When 14 was applied in the allylic amination of crotyl acetate using piperidine in the CFMR a similar rapid deactivation of the catalyst was observed. Dendritic catalyst 14-(Pd[allyl]Cl)12 was found to be stable according a retention measurement in dichloromethane as all the palladium was in the reactor after flushing the reactor with 10 times its volume. Also the addition of one or more equivalents of diethylamine, which converts (after nucleophilic attack) Pd(II) into Pd(0), did not result in leaching of palladium. Clearly, the catalyst decomposed under catalytic conditions only.
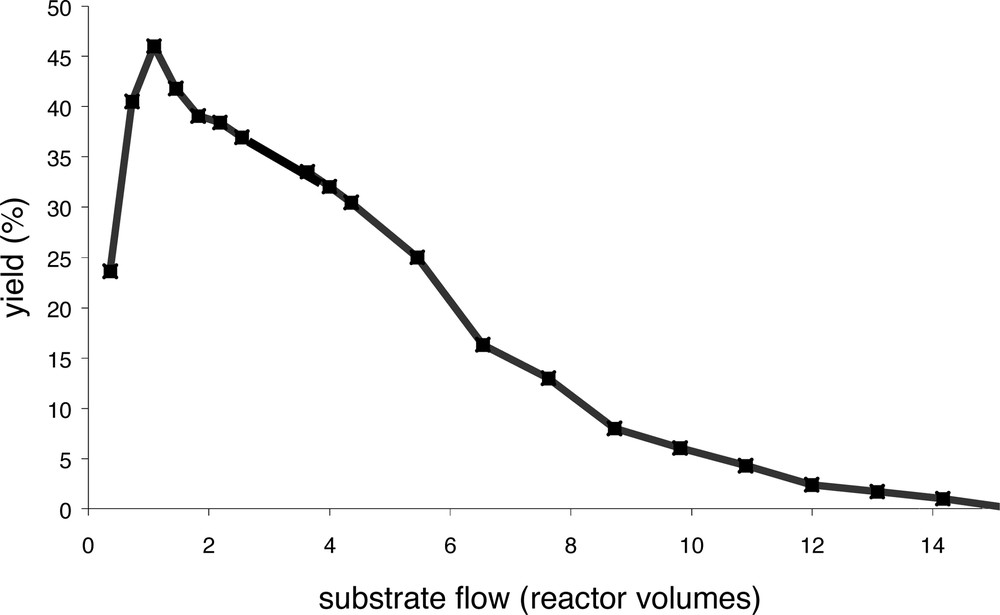
Application of dendritic ligand 14 in the continuous allylic alkylation of allyl trifluoroacetate and sodium diethyl 2-sodio-2-methylmalonate in a membrane reactor.
Dendrimer 11 was also applied as a ligand in the continuous allylic amination reaction. Fig. 9a shows that the maximum yield is reached after approximately five reactor volumes of substrate solution have been pumped through the reactor. This dendritic catalyst clearly is much more stable then 14 and during the next ten reactor volumes the formation of product was fairly constant. After this period the conversion was still more than 70% of the maximum reached, a decrease that corresponds to a retention of the dendritic catalyst of more than 98%. Under similar conditions with a P/Pd ratio of 4 instead of 2 (Fig. 9b) the dendritic catalyst was more active and also the stability had increased. The slight decrease in yield during this experiment is completely attributed to depletion of very small amounts of dendritic catalyst. Based on the curve in Fig. 9b the retention of the dendritic complex is estimated to be 98.5–99%, which is in the range of the expected values. It is interesting to note that the stability of the palladium catalyst is sensitive to relatively small changes of the dendritic backbone.

Application of dendrimeric ligand 20 in the allylic amination of crotyl acetate and piperidine using a CFMR (yield in% conversion of crotyl acetate in the product stream, (a) P/Pd = 2, (b) P/Pd = 4.
3.2 Core-functionalized dendrimers
The dendritic ferrocenyl ligands gave active palladium catalysts in the allylic alkylation reaction using cinnamyl acetate as the substrate and diethyl 2-sodio-2-methylmalonate as the nucleophile [50]. In Table 5 the average activity after 60 min and the selectivity for the trans product at full conversion for the various ligand systems are presented. All reactions reached full conversion within 24 h. The selectivity of the reaction was independent of the conversion and only the linear trans and the branched product were observed. Interestingly, both the selectivity and the activity changed with the size of the dendritic ligand used.
Reaction rates and regioselectivities of the palladium-catalyzed allylic alkylation of cinnamyl acetate using various ligandsa
| Ligand | TOF after 1h (102 mol (mol Pd)–1 h–1) | % Trans product (full conversion) |
| — | 0.2 | 96 |
| dppf (17) | 7.1 | 90 |
| dtpf | 9.3 | 89 |
| 18 | 7.5 | 87 |
| 19 | 5.7 | 87 |
| 20 | 1.7 | 79 |
| 21 | 2.9 | 84 |
The regioselectivity of the alkylation of cinnamyl acetate changes on going from dppf, dtpf and 18a to higher generation dendrimers 19a and 20; more branched product is formed when larger dendritic ligands are applied. This dendritic effect on the regioselectivity originates from the change in local polarity due to the size of the apolar dendritic shell. This was substantiated by using several solvent mixtures with different polarity in the allylic alkylation of cinnamyl acetate with diethyl 2-sodio-2-methylmalonate using dppf as the ligand giving a similar correlation between the regioselectivity and the solvent polarity. The dendritic complexes exhibit lower activities than the parent ligands dppf and dtpf, and the activities decrease in the series from lower to higher generation numbers (Table 6). This is in contrast to what would be expected in an apolar environment such as the interior of carbosilane dendrimers. The lower activities are attributed to the lower accessibility of the catalytic site located at the core of the dendrimer.
Results of the Pd-catalyzed allylic amination of crotyl acetate and piperidine, comparing the supramolecular dendritic catalyst with the monomera
| Catalyst | P/Pd | Conversionb (%) | trans (%) | cis (%) | Branched (%) |
| {Ester(32)}2Pd(crotyl)Cl a | 2 | 89 | 38 | 14 | 48 |
| {Acid(33)}2Pd(crotyl)Cl-dendrimer a | 2 | 91 | 37 | 15 | 48 |
| {Ester(32)}Pd(crotyl)Cl c | 1 | 80 | 33 | 6 | 61 |
| {Acid(33)}Pd(crotyl)Cl-dendrimer c | 1 | 72 | 33 | 6 | 61 |
3.2.1 Allylic alkylation in a continuous-flow membrane reactor
To show the applicability of core-functionalized dendritic catalysts in a continuous-flow membrane reactor, the third-generation dendritic ligand 20 was tested in a continuous allylic alkylation reaction. A THF solution of 20 and crotyl-palladium chloride dimer was mixed at room temperature and then injected into the membrane reactor. Subsequently, the reactor was fed with a solution of allyl trifluoroacetate, diethyl 2-sodio-2-methylmalonate and decane as internal standard (Fig. 7).
Fig. 10 shows that the conversion increases rapidly within the first 30 min (with a flow rate of 50 ml h–1 and a reactor volume of 20 ml, this is equivalent to 1 reactor volume) until it stabilizes around 45%, after which it remains almost constant for at least 8 h (the increase in conversion after 3.5 h is due to a temporary pump failure). The conversion after 8 h is similar to the initial conversion, which shows that the core-functionalized dendritic catalyst is very stable. This is in contrast with surface-functionalized dendritic catalysts 14 that showed considerable deactivation in time under similar conditions (Fig. 8). The encapsulation of the catalytic site within the dendritic sphere sufficiently protects the active species against deactivation (e.g., by membrane material, metal clustering with other metals). As shown above, the dendritic encapsulation is also responsible for the lower activity per catalytic centre, while in periphery-functionalized dendrimers the catalytic sites remain fully accessible. Dendritic architectures that have multiple catalytic sites located between the core and the periphery might combine a high stability with a high activity.
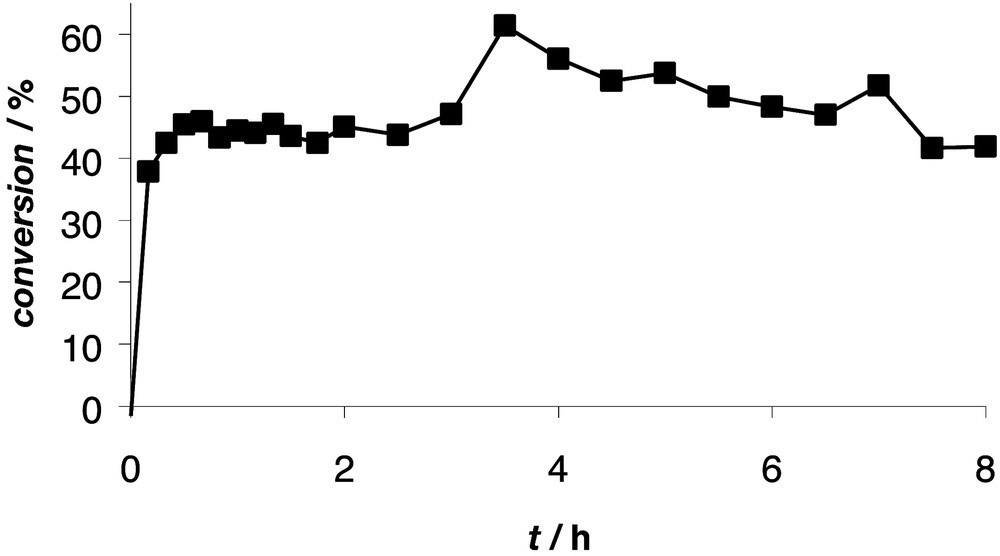
Allylic alkylation of allyl trifluoroacetate and diethyl 2-sodio-2-methylmalonate in a continuous-flow membrane reactor using dendritic ligand 20.
4 Non-covalently functionalized dendrimers as recyclable catalysts
So far we have used covalently functionalized dendrimers, which are accessible only after tedious synthetic procedures. An interesting alternative approach is the non-covalent anchoring of the catalyst to the dendrimer support using well-defined binding sites. This leads to multifunctional dendritic support that can be functionalized by addition of catalysts with proper binding motives. The reversible nature of this type of non-covalently anchoring allows controlled de- and re-functionalization of the support, which enables the reuse of the support and simplifies the variation of catalyst loading even during catalysis.
To achieve the supramolecular anchoring of catalysts, a soluble support must contain well-defined binding sites to which the tailor-made transition-metal catalysts with a complementary binding motive can be assembled (Fig. 11). To this end, we have used the fifth-generation poly(propylene imine) dendrimer functionalized with urea adamantyl units at the periphery (31) [52] that allows the non-covalent anchoring of phosphine ligands with the complementary binding motive [53] (Fig. 12).
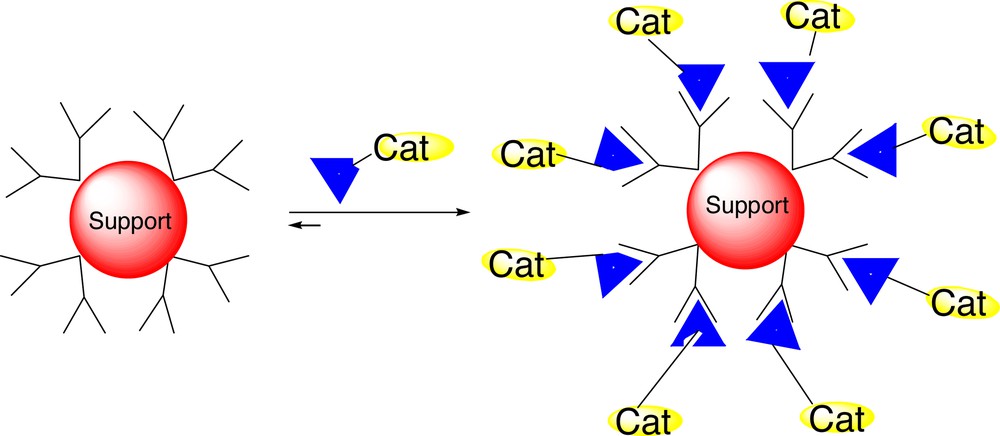
Schematic presentation of supramolecular anchoring of catalysts to dendritic support.
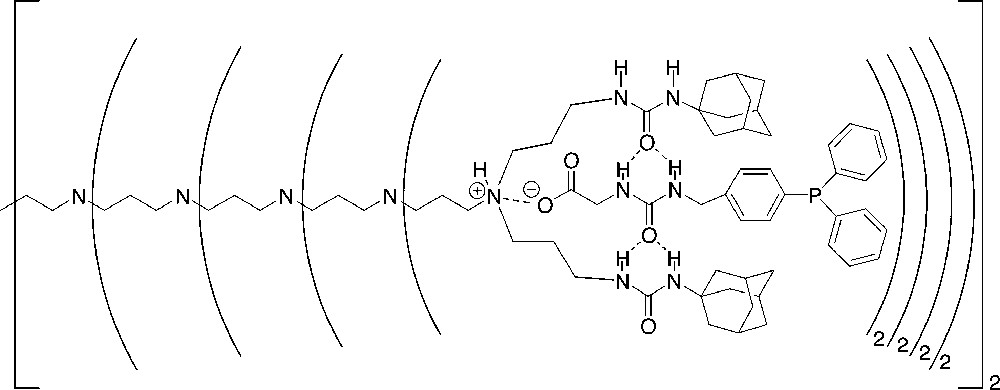
Phosphine ligands assembled to the periphery of a urea adamantyl functionalized poly(propylene imine) dendrimer (31).
The binding of the guest molecule into the periphery of 33 was studied by NMR spectroscopy showing that the guest molecule is positioned in the periphery of the dendritic host in a well-defined way (Fig. 12). In contrast to ester ligand 32, acid ligand 33 showed a strong affinity for the binding motive of the dendrimer (Fig. 13).
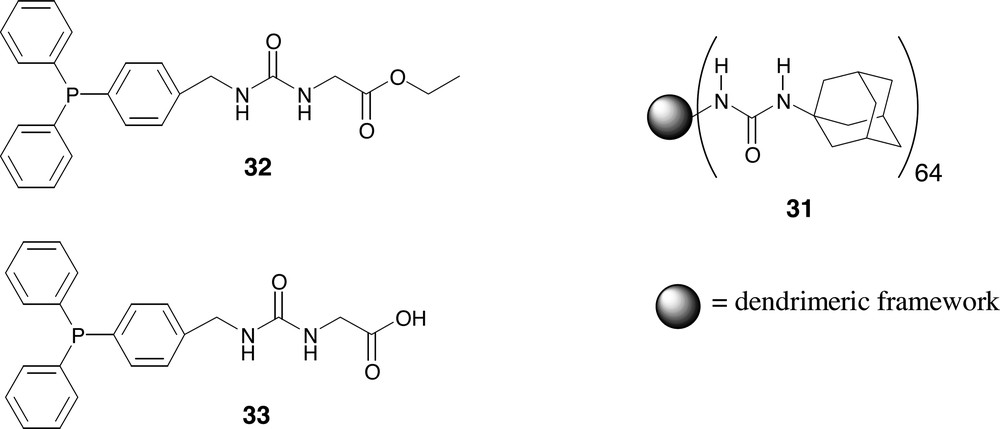
Phosphine containing guest molecule (33) binds in the binding site of the urea adamantyl-functionalized dendritic host (31), whereas the ester analogue 32 does not.
Pd-functionalized dendrimers were prepared by mixing all components, i.e. the dendrimer, a palladium precursor and 33. The obtained [{acid(33)}Pd(crotyl)Cl]32-dendrimer complex was characterized with 31P- and 1H-NMR. The [{acid(33)}2Pd(allyl)Cl]16-dendrimer complex remained intact after size exclusion chromatography (SEC) indicating that the binding constant of the acid(33)-PdCl(allyl)-complex to the periphery of the dendrimer as well as that of the Pd to the ligand is very high.
4.1 Catalysis using the supramolecular complexes
4.1.1 Batch process
The dendritic host containing 32 phosphine ligands assembled to the periphery of 31 was used as a multidentate ligand in the Pd-catalyzed allylic amination using crotyl acetate and piperidine as the substrate molecules. The reaction was performed under various conditions using [{acid(33)}2Pd(crotyl)Cl]16-dendrimer (P/Pd = 2) and [{acid(33)}Pd(crotyl)Cl]32-dendrimer (P/Pd = 1) as a catalyst. The reaction appeared to be fast; using the [{acid(33)}2Pd(crotyl)Cl]16-dendrimer (P/Pd = 2) and a substrate/palladium ratio of 100 the conversion was 91% after 5 min. Approximately the same rate was observed for the Pd-complex of ester(32) in the absence of the dendrimer (Table 6). This indicates that every active site on the dendrimer acts as an independent catalyst. These experiments show clearly that the supramolecular anchoring of the catalysts does not decrease the activity, in contrast to the general observation for catalysts immobilized on an insoluble support. Moreover, the product selectivity generated by the {acid(33)}2-Pd-dendrimer complex is the same as that induced by the {ester(32)}2-Pd-complex. The non-covalently functionalized dendritic catalyst used in these batchwise reactions was recycled using SEC. The conversion measured after one hour, however, was lower in the second run, suggesting that the catalyst has partly decomposed during the recycling procedure.
The use of the [{acid(33)}Pd(crotyl)Cl]32-dendrimer complex (P/Pd = 1) as a catalyst resulted in a similarly fast reaction (Table 6). Again the results for the {acid(33)}Pd(crotyl)Cl-dendrimer complex are similar to those for the {ester(32)}Pd(crotyl)Cl complex.
4.1.2 Application in the continuous-flow membrane reactor
The retention measured for the ester complex (32)Pd(crotyl)Cl (MW = 617.4) in the presence of the host-dendrimer is 97%, which is low compared to the supramolecular acid-dendrimer complex [(33)Pd(crotyl)Cl]32-dendrimer (99.4%). Interestingly, the use of a lower palladium loading (P/Pd = 2) increased the retention to 99.9%, suggesting that the palladium diphosphine complexes are bound more strongly due to cooperative effects. These results indicate that this type of supramolecular complexes will be efficiently separated from smaller compounds such as the reactants and products in a continuous-flow membrane reactor. The application in catalysis was studied using the allylic amination reaction.
In Fig. 14, the conversion is plotted as a function of the substrate flow. The conversion has increased to its maximum (ca. 80%) after approximately 1 h (which is equivalent to 1–2 reactor volumes of substrate solution pumped through the reactor). The conversion remains fairly constant during the first ten hours of the experiment. A small decrease is observed, which is presumably the result of slow deactivation of the catalyst, which has also been observed using covalently functionalized dendrimers. Upon using ester(32) the activity of the catalytic system drops more rapidly, since ester 32 is not bound to the dendrimer and is washed out of the reactor (retention = 97%). In this experiment slow deactivation of the catalyst occurs as well. These experiments clearly demonstrate that the non-covalently functionalized dendrimers are suitable as soluble supports for catalysts. These supramolecular supports can be conveniently reloaded with new catalysts, in contrast to their covalent analogues.
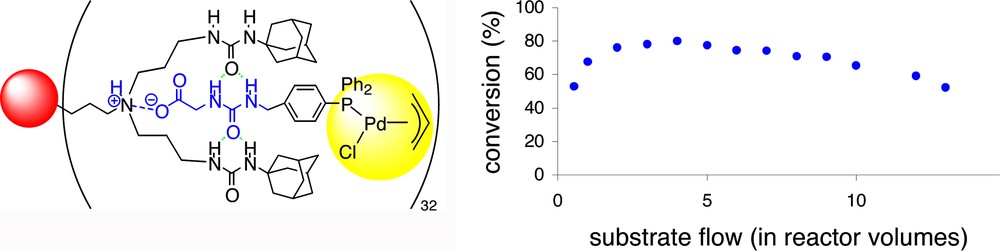
Application of non-covalently functionalized dendrimers (left) in a continuous-flow membrane reactor using acid (33) as a ligand.
5 Summary and outlook
We have studied both periphery- and core-functionalized dendrimers in the hydroformylation and allylic substitution reaction. The position of the catalytic sites and their spatial separation are determined by the geometry of the dendrimer and are very important for the performance of the catalyst. In the hydroformylation reaction we observed that the high local concentration of phosphine ligands in periphery functionalized systems can lead to slower catalysts, but also higher selectivities have been obtained. In the core-functionalized systems the selectivity and activity is largely determined by the ligand backbone located in the core. Moreover, the larger core-functionalized dendritic catalyst can be used for size selective catalysis. In the allylic substitution reaction we found that periphery functionalized systems have activities comparable to those of monomeric systems, but application in a continuous-flow membrane reactor showed that these catalyst systems can be very instable. In contrast, the core-functionalized dendrimers gave slower but very stable catalysts. Moreover, using these catalysts the selectivity was controlled by the apolar microenvironment created by the dendrimer. The precise tuning of the microenvironment (polarity, steric constraints) is still a difficult task, but it might provide a tool to enhance the selectivity of a reaction.
One of the limitations in dendritic catalysis is the troublesome synthesis of functionalized dendrimers, due to the required quantitative coupling of ligands to the periphery. The non-covalent strategy shown here leads to systems that are easy to functionalise and can be reused to function as support for new catalyst. Employing the concept of non-covalent anchoring simplifies the route towards sophisticated dendritic catalysts since ligand modification with the binding motive is straightforward. Moreover, this approach opens the way towards the use of multipurpose supports, not only for dendrimers but also other supports, which can be functionalized and refunctionalised with multiple complex catalytic systems containing a relatively simple binding motive.