

1 Introduction
The synthesis of organic-inorganic hybrid materials is based on the association of organic and inorganic moieties in order to obtain a new material with the desired properties (mechanical, electric, optical, textural, catalytic, etc.) [1–3]. Judeinstein and Sanchez [1] proposed to distinguish two main classes of hybrid materials based on the nature of the interaction between organic and inorganic parts. In Class-I hybrids, the organic part is trapped in the inorganic matrix or bound by weak interactions, whereas in Class-II hybrids the organic and the inorganic parts are linked by strong chemical bonds.
Class-II hybrids can be obtained not only by sol–gel processing but also by surface modification of an inorganic support. In surface modification, the term ‘coupling agent’ originally stood for surface modifiers bearing reactive organic groups (vinyl, amino, etc.) allowing the formation of stable bonds between an organic polymer and a mineral. By extension, the term coupling agent is now often used for surface modifiers bearing non-reactive organic groups (e.g. long alkyl chains). Silane coupling agents such as RSiX3 (X = Cl, alkoxy) are the best-known coupling agents. In sol–gel processing, the term coupling agent is not customary; organoalkoxysilanes are called precursors or monomers, complexing ligands such as carboxylic acids or β-diketones are called modifiers. Here, we propose to call ‘coupling molecules’ the molecules allowing the anchoring of organic groups (reactive or non-reactive) to an inorganic surface (surface modification) or to an inorganic network (sol-gel processing) as illustrated in Fig. 1.

General representation of organic–inorganic Class-II hybrids prepared by surface modification of an inorganic support (top) and by sol–gel processing (bottom).
According to this definition, all Class-II hybrids involve coupling molecules. Note that in the particular case of sol–gel derived polysilsesquioxane hybrids (RSiO1.5)n no inorganic precursor is used, the inorganic Si–O–Si bond network resulting from the polycondensation of the RSiX3 molecules.
Organosilane coupling molecules such as organotrialkoxysilanes RSi(OR′)3 are ideally suited to the design of Class-II hybrid materials derived from silicon-containing inorganic phases (silica, silicate glasses, silicon nitride, etc.); in this case, the bonding results from the facile formation of strong Si–O–Si bonds between the inorganic phase and the organosilane.
In our group, we are interested in developing the use of organophosphorus acids (phosphoric, phosphonic, and phosphinic) and their derivatives (salts, esters) as coupling molecules. These organophosphorus compounds, of general formula RxP(O)(OX)3–x (x = 1 or 2, X = H, Na, alkyl, etc.) appear complementary of organosilanes: they show an excellent affinity toward metal and transition metal containing supports (oxides, hydroxides, carbonates, phosphates, etc.). The ease of formation of M–O–P bonds is well illustrated by the many examples of metal phosphates or phosphonates reported in the literature. Layered metal (IV) phosphates and phosphonates have attracted much attention for their potential applications as ion exchangers, proton conductors, and catalysts [4–7]. Unfortunately, layered metal phosphonates are not intrinsically porous, and only the organic groups exposed at the surface of the particles are accessible.
In this ‘mini-account’, we wish to describe the various approaches that we explored to improve the accessibility to the organic groups in hybrid materials based on organophosphorus coupling molecules.
In the first part, we present the synthesis of microporous pillared zirconium phosphonates (Fig. 2a) using an original sol–gel route involving the non-hydrolytic condensation of P–OH groups with a metal alkoxide. Note that in such compounds the inorganic part is built of M–O–P bonds only, with a M/P ratio of 1/1/2, as in α-zirconium phosphate or phosphonates.
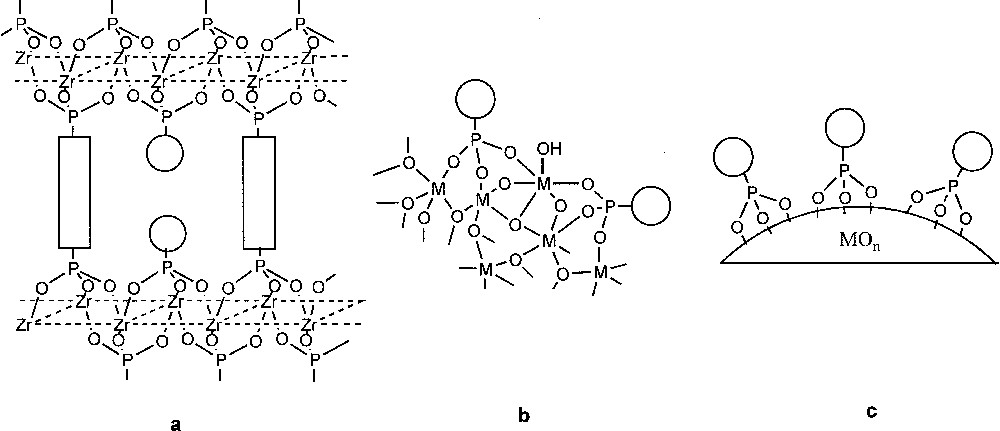
Schematic representation of a microporous pillared phosphonate of zirconium (a), a metal oxide/phosphonate hybrid prepared by sol–gel (b), and a metal-oxide nanoparticle modified by phosphonate groups (c).
In the second part, we describe the preparation of new metal oxide-phosphonate hybrid solids by a two-step sol–gel route (Fig. 2b). Whereas sol–gel processing of organoalkoxysilane precursors has been extensively used in the last 20 years to prepare Class-II hybrids, it is only very recently that a sol–gel route using organophosphorus coupling molecules was proposed [8, 9]. These hybrid solids may be seen as a porous metal oxide phase homogeneously modified by dispersed phosphonate (or phosphinate) groups. Here the inorganic network is built of M–O–M and M–O–P bonds, and the M/P ratio can be varied between about 1 to 10 or more.
In the third part, we present our work on the surface modification of non-porous metal oxide nanoparticles (Fig. 2c) by phosphonic and phosphinic acids and more particularly by their ethyl ester and trimethylsilyl ester derivatives, which had not been used before as coupling molecules. The M/P ratio in the resulting hybrid solids may range between about 10 to 100 or more, depending on the specific surface area of the support and the density of grafting. Finally, in the last part we will illustrate the potentialities of the sol–gel and surface modification approaches with examples of the immobilization of organometallic metal complexes using phosphine-phosphonate coupling molecules.
2 Sol–gel route to microporous pillared phosphate–phosphonate of zirconium
Since the initial work of Dines et al. [10], the creation of microporosity in metal phosphonates by alternating long bridging organic groups (‘pillars’) and small functional pendent groups (Fig. 2a) has been investigated by several groups [11, 12]. Ideally, the dimension of the interlayer micropores should depend on the pillaring fraction and on the dimensions of the pillars and of the pendent groups. However, such mixed compounds are metastable, and phase segregation was often observed when they were prepared by precipitation in aqueous medium and refluxing in concentrated HF solution to obtain better-crystallized products. A few years ago, we proposed to prepare these materials by a sol–gel procedure, based on the non-hydrolytic condensation between an organophosphorus acid and a metal alkoxide (Eq. (1)) [13]. The same condensation reaction was initially used to prepare α-zirconium phosphate [14]. In addition, to improve the crystallinity of our samples while avoiding phase separation, we used a mild hydrothermal treatment without HF.
We prepared a series of mixed phosphate–biphenylenebis(phosphonate) of zirconium, with a pillar fraction ranging from 0 to 1 (Eq. (2)).
The solids obtained were then annealed under hydrothermal conditions (110 °C for 7 days) to improve their degree of crystallinity. The powders obtained after drying consisted of aggregated primary particles with a size of about 20 nm. These primary particles were found to be crystalline, with a layered structure and an interlayer distance of 1.4 nm, consistent with the biphenylene pillars used.
The variation of specific surface area and micropore volume with the pillar fraction (Fig. 3) demonstrated that the high specific surface area of the mixed samples (x = 0.2 to 0.6) originated from the presence of interlayer micropores. The low surface area and micropore volume found for the sample with a 0.8 pillar fraction can be ascribed to the high probability of forming closed micropores at high pillar fractions. In addition, the distribution of micropore diameter (Fig. 4) showed the presence of well-defined micropores in the mixed samples [13]. It is worth noticing that the micropore distribution found in our mixed samples was very similar to the one obtained by Alberti and co-workers using their ‘pillars with base’ approach [15]. In addition to this intraparticle microporosity, our mixed samples exhibited significant interparticle mesoporosity (100–180 m2 g–1) related to the very small particle size.

Variation of specific surface area and micropore volume with the fraction of pillars x.

Micropore size distribution for different pillar fractions x.
Despite these encouraging results, we did not pursue this approach. Indeed, we found that it was not really possible to control the dimensions of the micropores, by playing on the pillar density or on the length of the pillar.
3 Sol–gel route to metal oxides modified by phosphonate or phosphinate groups
Synthesis of organic-inorganic hybrids by sol–gel processing offers many possibilities: control of the organic content, of the surface area and porosity, and shaping of the final material before the liquid-to-gel transition (film coating for instance). Recently we have reported the incorporation of organophosphorus groups within zirconium, titanium and aluminium oxides by a two-step sol–gel processing [8, 9, 16].
As shown in Fig. 5, a metal alkoxide precursor was first mixed with a solution of phosphonic or phosphinic acid in an organic solvent; P–O–M bonds form at this stage by the same condensation reaction as the one used in the former part (Eq. (1)). Then neutral water was added, leading to the formation of the M–O–M bonds of the oxide network by hydrolysis/condensation of the residual alkoxide groups. Alternatively, the acid precursors may be replaced by their silyl ester derivatives (X = SiMe3 in Fig. 5), which are more soluble in organic solvents.

2–Step sol–gel route to metal oxide/phosphonate (x = 1) or phosphinate (x = 2) hybrid solids.
A detailed study was carried out with Ti(OiPr)4, using diphenyl phosphinic (DPPA) and phenyl phosphonic acids (PPA) [17]. These organophosphorus acids act as chemical modifiers of the metal alkoxide [18]. Depending on the P/Ti and H2O/Ti ratios, and on the nature of the organophorus acids, molecular species, sols, gels, or precipitates were obtained. For P/Ti ratios ranging from 0.25 to 1, and for H2O/Ti ratios close to the ratio theoretically necessary to completely condense all the remaining alkoxide groups, gels were obtained in the case of PPA, whereas stable sols formed with DPPA. Whatever the H2O/Ti ratio, the P/Ti ratio was retained, showing that there was no removal of the PPA or DPPA ligands upon hydrolysis.
For low H2O/Ti ratios, soluble, molecular species are obtained: oxo-alkoxide clusters modified by PPA or DPPA, such as [Ti4(μ3–O)(OiPr)5(μ-OiPr)3(PhPO3)3]·THF (1) and [Ti(O)(OiPr)(O2PPh2)]4 (2) (theoretical H2O/Ti ratios of 0.25 and 1, respectively; Fig. 6), which were characterized by single crystal X-ray structure analysis and NMR spectroscopy [19–21].

Molecular structure of [Ti4(μ3–O)(OiPr)5(μ-OiPr)3(PhPO3)3]·THF (1) and [Ti(O)(OiPr)(O2PPh2)]4 (2).
Based on solution NMR spectroscopy, compound 1 was the major product at the beginning of the hydrolysis step (whatever the Ti/P ratio in the range 2–10). Its structure highlighted that condensation reactions between P–OH and Ti–OR groups were completed at this stage, and that phosphonate groups behaved as tridentate ligands linking three different titanium atoms. Moreover, the hydrolysis of 1 led to a gel (Ti/P = 1.33) whose 31P NMR MAS spectrum was similar to that of gels stemming from Ti(OiPr)4/PPA mixtures; accordingly, it can be regarded as a plausible intermediate in the sol–gel processing [22]. Conversely, compound 2 was found to be relatively stable toward hydrolysis (the hydrolysis reaction was not completed after 24 h in the presence of excess water at room temperature); compound 2 likely corresponds to a secondary product rather than to an intermediate.
Phenylphosphonate-containing gels were also prepared from zirconium and aluminium alkoxides [8, 16]. From solid-state 31P MAS NMR spectroscopy, XRD, and EDX analyses, the xerogel samples could be described as a homogeneous dispersion of phosphonate groups within an amorphous metal oxide matrix. In the case of TiO2 gels the phenylphosphonate groups were shown to be strongly anchored; refluxing samples in aqueous solutions of a pH ranging from 0 to 10 for 12 hours resulted in less than 2% loss of phosphorus groups. Phase separation into titanium dioxide and titanium phenylphosphonate only occurred under severe conditions (heating for 7 days at 120 °C at pH 0) [16].
It is worth noticing that a one-step sol–gel method (Fig. 7) was used by Bujoli and co-workers to immobilize rhodium and iridium 2,2′-bipyridine complexes onto in situ-generated titanium dioxide particles, to prepare supported catalysts for hydrogenation reactions under dihydrogen pressure [23]. The excellent activities of the resulting supported catalysts compare well with the homogeneous parent systems; no significant metal leaching was observed on recycling.
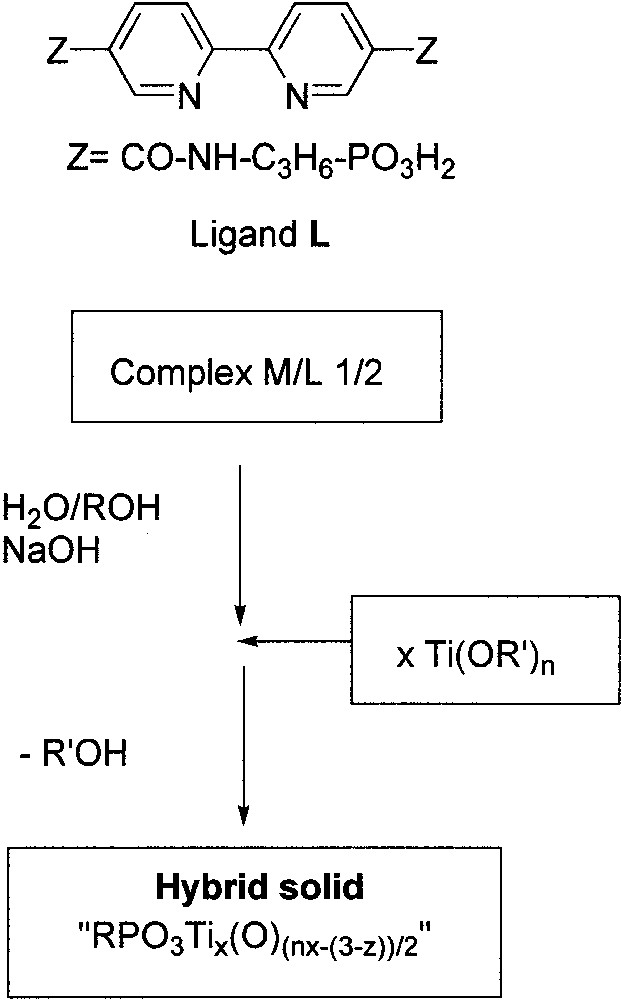
One-step route used for the immobilization of rhodium and iridium 2,2′–bipyridine complexes onto in-situ generated TiO2 particles [23].
In conclusion, the use of organophosphorus precursors in sol–gel processing is very attractive for the preparation of organic-inorganic hybrid materials based on metal oxides. The advantages of the resulting hybrid materials, over those derived from organosilicon precursors, are their high level of homogeneity (self-condensation between P–OH groups does not occur under the sol–gel conditions, whereas self-condensation between Si–OH groups may lead to the formation of separate domains) and, in the case of TiO2 or ZrO2, their high stability, especially toward basic hydrolysis.
4 Surface modification of metal oxide nanoparticles
Organophosphorus coupling molecules, such as phosphonic acid, monoalkylphosphoric acid, and their salts, have been used to modify the surface of metal oxides for various applications [24–29].
In our group, we have studied the surface modification of titania [30] and alumina [16, 31] nanoparticles by model organophosphorus coupling molecules: phenylphosphonic and diphenylphosphinic acids, their ethyl esters, and their trimethylsilyl esters. One important result of these studies was to demonstrate that organic-soluble phosphonic and phosphinic ester derivatives could be used to modify surfaces. FTIR and 31P MAS NMR spectra indicated that the bonding mode to the surface of acids and the related esters was similar.
For example, the FTIR spectra of titania nanoparticles modified by PhPO3H2, PhPO(OSiMe3)2, and PhPO(OEt)2 were nearly identical in the 900–1300 cm–1 region corresponding to the vibrations of the phosphonate group (Fig. 8). In all cases, the disappearance of the P=O stretching band and of the vibrations typical of P–OH, P–OSiMe3, or P–OEt groups suggested that the main bonding mode to the surface is tridentate (PhP(OTi)3 sites) as in layered titanium phosphonates, molecular alkoxophosphonates [21], or TiO2–PhPO3 hybrids obtained by sol–gel processing [9]. These tridentate sites apparently result from the coordination of the phosphoryl oxygen to surface Lewis acid sites and from the condensation of P–OH, P–OSiMe3, or P–OEt groups with Ti–OH surface groups, according to Eq. (3).

FTIR (DRIFT) spectra of TiO2 nanoparticles (P25 Degussa) modified by PhPO3H2 (72 h at RT in MeOH/H2O), PhPO(OSiMe3)2 (24 h at RT in CH2Cl2), and PhPO(OEt)2 (24 h at 40 °C in CH2Cl2). All powders were dried at 120 °C under vacuum.
The reactivity of the silylated ester, PhPO(OSiMe3)2, is not surprising, as the PO–Si bonds are easily cleaved by water or alcohol. On the other hand, P–O–C bonds in PhPO(OEt)2 are not easily hydrolysed (unlike Si–O–C bonds) and their cleavage on an oxide surface suggests that the coordination of the phosphoryl oxygen to the surface assists the condensation by increasing the electrophilicity of the P atom (Fig. 9).
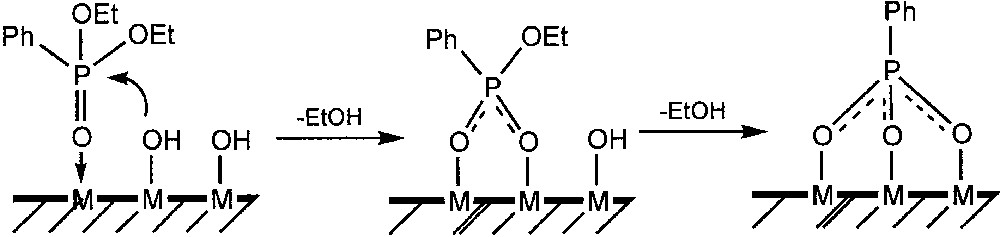
Schematic representation of the reaction of PhPO3Et2 on a metal-oxide surface, leading to a tridentate PhP(O3Ti)3 species.
In the case of organosilane coupling molecules, the quality of surface modification is very sensitive to the amount of water in the solvent or adsorbed on the surface of the oxide; this behaviour is related to the self-condensation involving Si–OH groups formed by hydrolysis (Fig. 10).

Schematic representation of the influence of water on the surface modification of an oxide surface by an organotrimethoxysilane.
An important advantage of organophosphorus acids is that condensation of P–OH or P–O– groups does not take place under mild conditions and that they are stable in aqueous medium. Hence, the quality of surface modification does not depend on the amount of water; actually, surface modification may easily be performed in water. In principle, as organophosphorus coupling molecules cannot self-condense by formation of P–O–P bonds, they can only react with the surface and should lead only to monolayers. However, depending on the chemical stability of the oxide and the reaction conditions (temperature, concentration, pH, nature of the solvent) a dissolution-precipitation process may compete with surface modification, as illustrated in Fig. 11 for phosphonic acid coupling molecules.
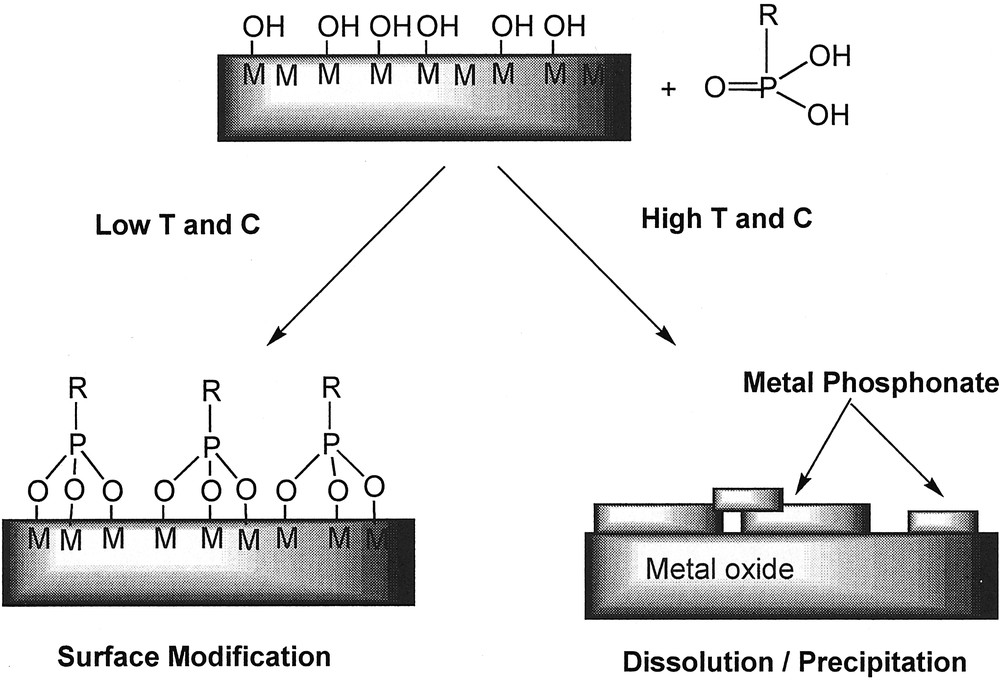
Competition between surface modification and dissolution–precipitation processes in the reaction between a metal oxide surface and a phosphonic acid.
Solid-state 31P MAS NMR proved particularly useful to evidence this dissolution-precipitation process. Even in the case of titania, known for its exceptional chemical stability, we found that layered titanium phenylphosphonate, Ti(PhPO3)2, could form under relatively mild conditions, such as refluxing in water in the presence of PhPO3H2 or at 120 °C in organic medium with PhPO3H2 and PhPO(OSiMe3)2 [9]. The presence of bulk titanium phenylphosphonate was evidenced by a sharp resonance at –4 ppm beside the broader signals around 12 ppm corresponding to the phenylphosphonate groups anchored to the surface (Fig. 12). It is noteworthy that only surface modification was observed in the case of PhPO3Et2, even under severe conditions.
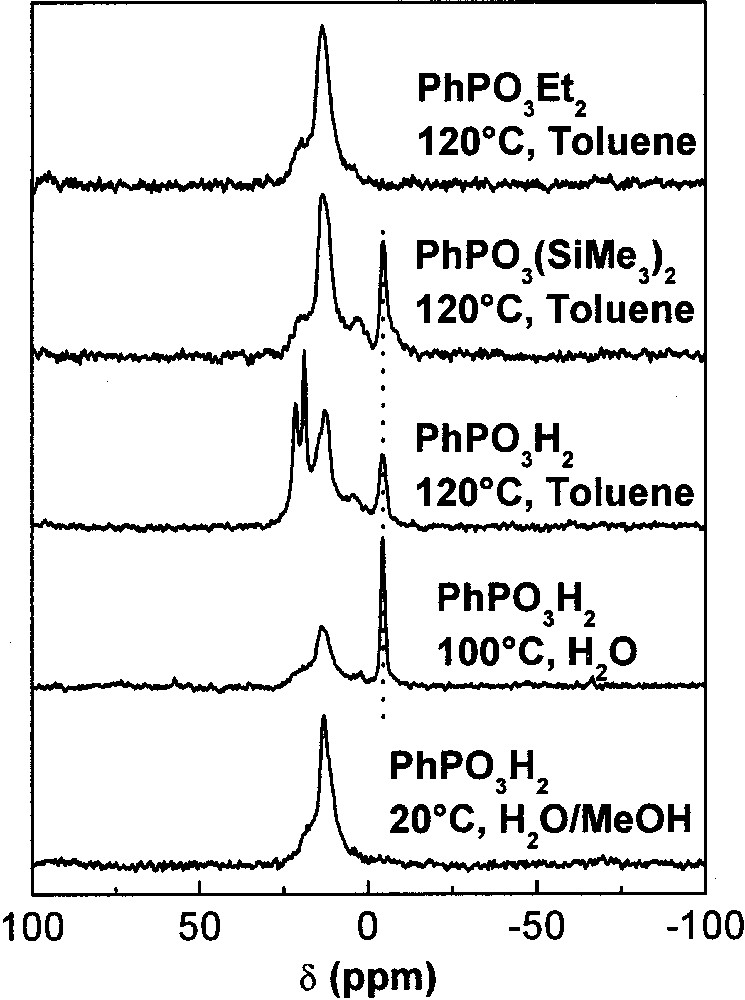
31P MAS NMR spectra of titania nanoparticles (P25 Degussa) modified by 1.56 × 10–3 mol of coupling molecule per g TiO2 under different conditions. From bottom to top: PhPO3H2 concentration 2.6 × 10–3 mol l–1, 3 days at 20 °C; PhPO3H2 concentration 39 × 10–3 mol l–1, 1 day at 100 °C; PhPO3H2 concentration 0.1 mol l–1, 1 day at 120 °C; PhPO(OSiMe3)2 concentration 0.1 mol l–1, 1 day at 120 °C; PhPO(OEt)2 concentration 0.1 mol l–1, 1 day at 120 °C. The dotted line indicates the position of the resonance at –4 ppm arising from layered titanium phenylphosphonate.
In view of the excellent stability and low solubility of TiO2, these results suggest that Ti–O–Ti bonds can be cleaved by PhPO3H2 and PhPO(OSiMe3)2 but not by PhPO3Et2. A mechanism was suggested, involving the transfer of a proton or a trimethylsilyl group (Fig. 13).
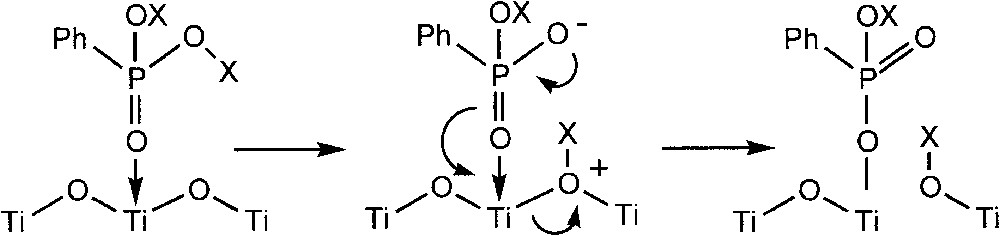
Schematic representation of the cleavage of Ti–O–Ti bonds by PhPO3H2 and PhPO(OSiMe3)2.
In the case of alumina nanoparticles, significant dissolution-precipitation was found even at room temperature with PhPO3H2 in water at pH 4 or in THF, as well as with PhPO(OSiMe3)2 in CH2Cl2. On the other hand, only surface modification was observed with PhPO3H2 in water at pH 6 or with PhPO3Et2 in CH2Cl2 at 40 °C.
In conclusion, esters of organophosphorus acids can be used to modify the surface of inorganic supports in organic solvents. It must be noted that alkylesters and trimethylsilylesters are intermediates in the synthesis of acids. The alkylesters are somewhat less reactive and lead to lower grafting densities than the acids or the trimethylsilylesters, but they appear much more selective, giving exclusively surface modification.
5 Application to the immobilization of organometallic complexes
The preparation of organic-inorganic hybrid heterogeneous catalysts is an important application of organosilane coupling agents. As phosphines form stable complexes with a great variety of transition metals, bifunctional phosphine-alkoxysilanes, such as Ph2P–(CH2)3–Si(OEt)3, have been extensively used to immobilize phosphines–metal complexes on silica supports [32]. However, it was recently shown that in practice these coupling molecules could not be used with other oxide supports, such as TiO2 and MgO [33]. Conversely, we were able to effectively immobilize phosphines and their Pd or Pt complexes on TiO2 and ZrO2 supports using bifunctional phosphine-phosphonate coupling agents [34].
In the field of catalysis, phosphonated phosphines have been used to prepare hemilabile transition metal complexes [35] and water-soluble complexes [36–38]. For our needs we prepared two diethyl phosphonated phosphine ligands of formula Ph2P(CH2)3PO3Et2 (ligand L) and Ph2P(4–C6H4PO3Et2) (ligand L′) (Eq. (4)).
Reaction of L and L′ with PtCl2(PhCN)2 and PdCl2 led to several complexes, five of which were characterized by single-crystal X-ray diffraction [39]:
- • cis-[Et2O3P–(CH2)3–PPh2]2PtCl2 3 (Fig. 14)
- • trans-[Et2O3P–(CH2)3–PPh2]2PtCl2.H2O 4
- • trans-[(Et2O3P–(CH2)3–PPh2) PdCl2]2 5
- • trans-[Et2O3P–(CH2)3–PPh2]2PdCl2.H2O 6
- • trans-[Et2O3P–C6H4–PPh]2PdCl2.2 CH2Cl2 7 (Fig. 15).
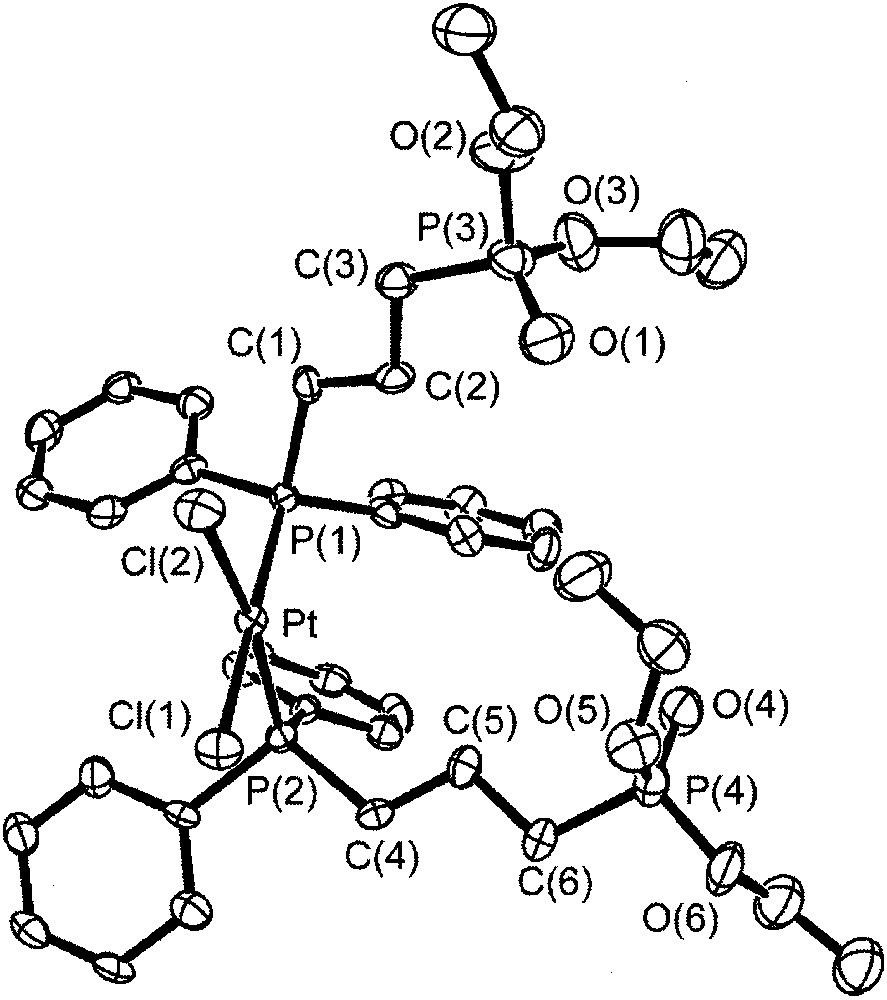
Structure of compound 3.

Structure of compound 7.
In all of the above compounds, complexation involves only the phosphine end; the strong polarization of the P=O bond is illustrated by the formation of hydrogen bonds with water molecules in 4 and 6, and with a CH2Cl2 molecule in 7. The length of the C–H···O hydrogen bond in the latter compound was exceptionally short, with a C···O separation 3.094(3) Å. These compounds were used as precursors and also as models in the 31P NMR characterization of the immobilized complexes.
There are two possibilities to prepare metal oxide gels modified by phosphine-metal complexes: the complexation of the phosphine groups can be done before or after the sol–gel processing step (Fig. 16). Diethylphosphonates cannot be used as sol-gel precursors. Thus, ligands and complexes were converted to silylesters by reaction with Me3SiBr [40]. It must be noted that the geometry of the complexes (cis or trans) was not modified by the silylation, as shown by 31P NMR.

Sol–gel routes used to immobilized phosphines and phosphine-metal complexes.
The 31P MAS NMR spectrum of a ZrO2 xerogel modified by O3P–(CH2)3–PPh2 groups shows the expected resonances corresponding to the phosphonate and phosphine groups (Fig. 17a). This sample was refluxed for 24 h in a solution of PtCl2(PhCN)2 in CH2Cl2, leading to the nearly complete disappearance of the phosphine groups and to new resonances between –5 and 20 ppm, ascribed to phosphine groups complexed to PtCl2 (Fig. 17b). This spectrum is quite similar to the spectrum of the xerogel prepared from cis–[(Me3SiO)2OP–(CH2)3–PPh2]2PtCl2, obtained from compound 3 by silylation (Fig. 17c). The immobilized complexes are probably in both cis and trans geometries: the 31P MAS NMR spectra show two resonances at 10.2 and –6.3 ppm in a 1:1 ratio for cis[Et2O3P–(CH2)3–PPh2]2PtCl2 (3) and a single resonance at 12.4 ppm for trans–[Et2O3P–(CH2)3–PPh2]2PtCl2·H2O (4).
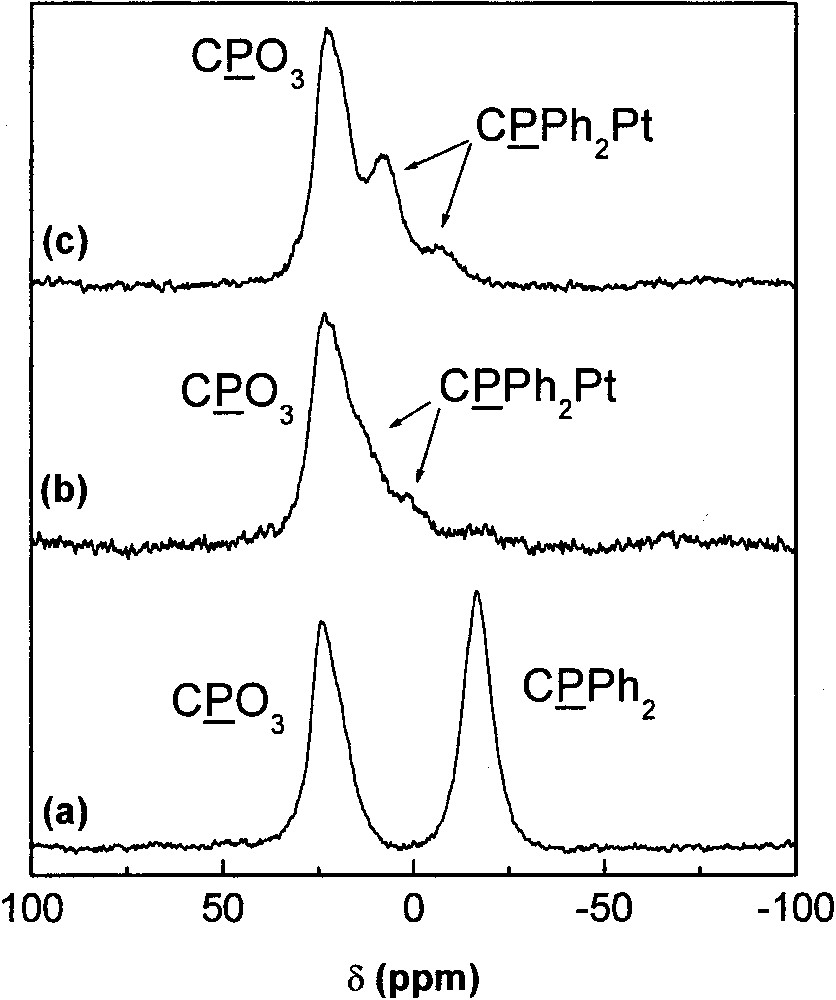
31P MAS NMR spectra of hybrid xerogels. (a) (Me3SiO)2OP–(CH2)3–PPh2 + 4.25 Zr(OiPr)4.iPrOH + 7.5 H2O; (b): sample (a) after complexation with PtCl2(PhCN)2; (c) cis–[(Me3SiO)2OP–(CH2)3–PPh2]2PtCl2 + 10 Zr(OiPr)4·iPrOH + 18 H2O
As mentioned in section 3, phosphonate diethylesters can be used to modify metal oxide surfaces. Thus, the complex trans-[Et2O3P–C6H4–PPh]2PdCl2·2 CH2Cl2 (7) was successfully grafted by reaction with titania nanoparticles at 40 °C in CH2Cl2 for 1 day, as shown by the 31P MAS NMR spectrum of the resulting powder (Fig. 18). The resonance at 12 ppm may be ascribed to the arylphosphonate groups linked to the surface, and the signal at 22 ppm to the phosphine groups coordinated to the Pd atoms, probably in a trans geometry. Indeed, the 31P MAS NMR chemical shift of phosphine groups in the starting trans complex is 21.8 ppm. Conversely, the low field resonance at 33 ppm may be tentatively ascribed to phosphine groups of immobilized complexes in cis geometry, indicating some isomerization. This resonance might also be ascribed to phosphine oxide groups, resulting from the decomplexation and oxidation of some phosphine ligands.
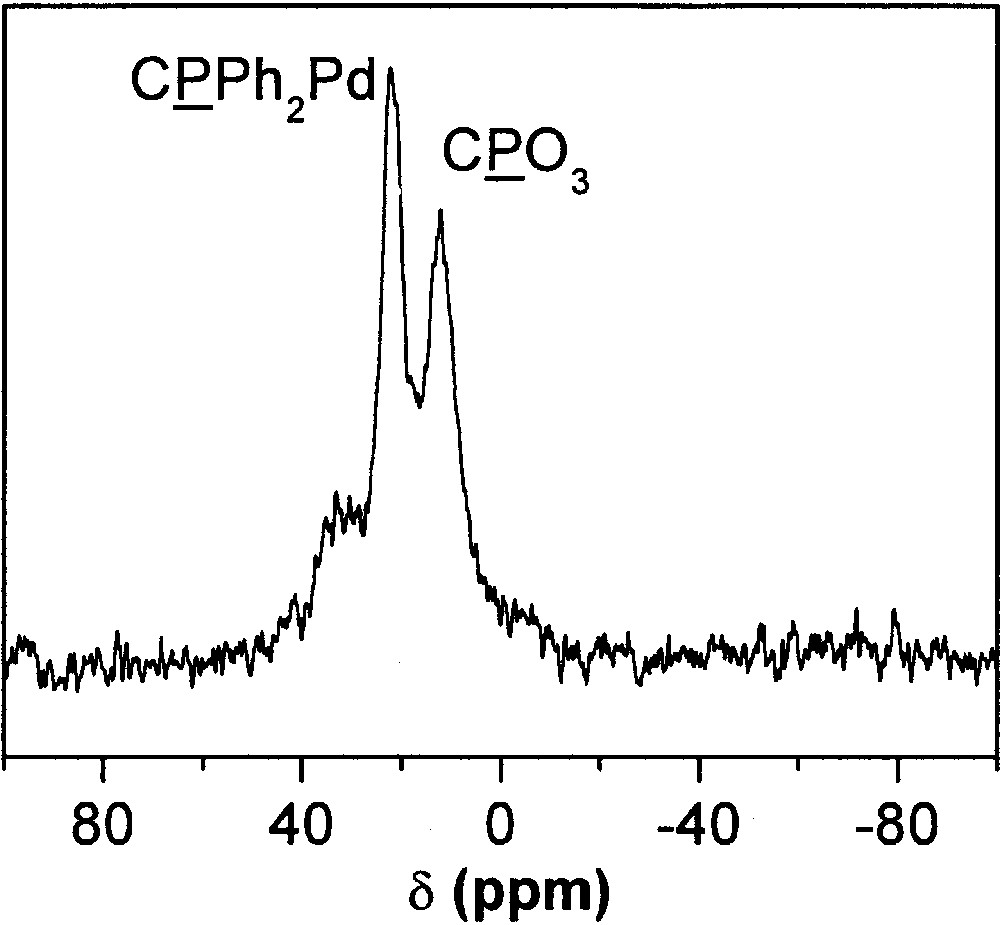
31P MAS NMR spectrum of titania nanoparticles (P25 Degussa) modified by trans-[Et2O3P–C6H4–PPh]2PdCl2·2 CH2Cl2 by reaction in CH2Cl2 at 40 °C for 1 day.
6 Conclusion
Organophosphorus acids (phosphoric, phosphonic, and phosphinic) and their derivatives (salts, esters) are highly promising coupling molecules, complementary of organosilanes. Organophosphorus derivatives are certainly the best alternative for the preparation of Class-II hybrid materials based on metal oxides, using either sol-gel processing or surface modification. Surface modification of various inorganic supports by these coupling molecules has already led to several applications in the fields of composite materials [41, 42], separation [24], catalysis [43], for the elaboration of photovoltaic [25, 44] and optical devices [45] or in the biomedical area [46, 47]. The sol–gel processing is still in its infancy, but there is no reason why it should not also find applications in the same fields.