Protecting groups play a key role in the synthesis of complex natural products [1]. This holds especially true for the synthesis of oligosaccharides, of which the monomeric carbohydrate building blocks usually contain up to five different hydroxyl functions and can also bear amino and carboxylate groups [2]. Discrimination of these functionalities requires a careful protecting group strategy and typically involves multistep protocols. Although protecting groups primarily function to mask a given functionality on the carbohydrate core, they also have a profound effect on the overall reactivity of a carbohydrate building block and can control the stereochemical outcome of a glycosylation reaction [2]. Furthermore, protecting groups can be used to introduce extra functionality on the carbohydrate core, such as visualization and/or purification handles. This review describes selected examples of new protecting groups and protection strategies in carbohydrate chemistry developed since the beginning of this century and highlights how protecting group chemistry has evolved from a necessary time consuming burden to a sophisticated synthetic tool for the efficient and stereoselective assembly of oligosaccharides.
1 Advances in the regioselective protection of carbohydrates
The regioselective manipulation of the different hydroxyl groups on a carbohydrate monomer is key to any protecting group strategy. Although the hydroxyl groups are of comparable reactivity, they can be discriminated by exploiting their subtle reactivity differences and their relative orientation [2]. The nucleophilicity of the different hydroxyls under neutral or acidic conditions increases from the anomeric to the secondary to the primary alcohol function. The reactivity difference between an axially and an equatorially oriented hydroxyl group can often be exploited to attain a regioselective protection step. Commonly, the use of cyclic protecting groups [2c] presents a more robust way to discriminate between the different functionalities on a carbohydrate ring. For example, benzylidene type acetals are widely used to selectively mask the C-4 and C-6 alcohols, isopropylidene ketals to block two neighboring cis hydroxyls, and butane 2,3-bisacetals to protect vicinal diequatorial diols. The anomeric hydroxyl group is most acidic and therefore selective protection (or deprotection) of the C-1-OH can be achieved in the presence of other secondary hydroxyls using basic reaction conditions. Furthermore, the hemiacetal function, encompassing the C-1 hydroxyl can selectively be manipulated to provide a (thio)acetal function. Most of the new anomeric O-acyl and O-alkyl groups that have been introduced the last decade not only mask the anomeric hydroxyl but also have the purpose to serve as an anomeric leaving group and therefore fall beyond the scope of this review [3].
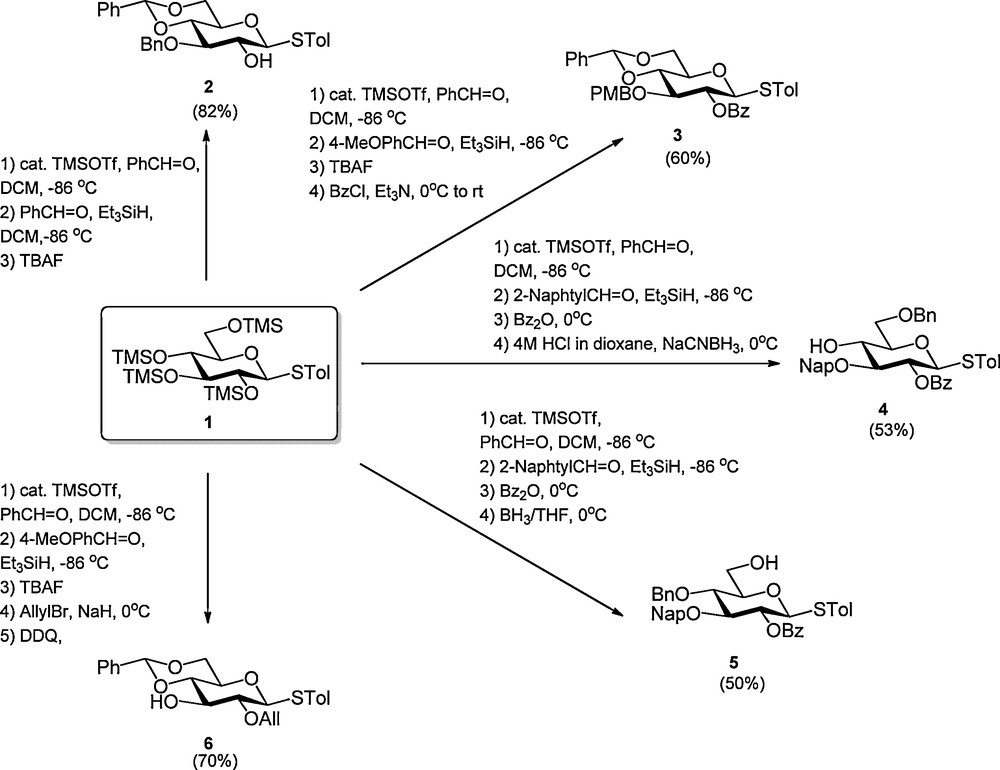
Recently several sequential procedures have been developed to streamline the regioselective protection of carbohydrates. Hung et al. disclosed that anomerically protected per-silylated carbohydrate monomers can be transformed into a wide array of differentially protected building blocks using a one-pot protocol, which combines up to five reaction steps (Scheme 1) [4]. Because all steps are consecutively executed in the same reaction vessel, the intermediate work-up and purification steps are omitted making this process highly efficient. The strategy builds on the trimethylsilyltriflate (TMSOTf) catalyzed installment of a C-4, C-6 arylidene function, which is followed by the regioselective formation of a benzyl type ether at C-3 [5]1. Next, the C-2-OH can be acylated and the arylidene opened to liberate either the C-4-OH or C-6-OH. Alternatively the C-3-benzyl ether is removed to expose the C-3-alcohol. Instead of the introduction of a C-2 acyl functionality also the incorporation of various ethers was described. Using the one-pot protocol, Hung et al. reported the synthesis of a large panel of differentially protected glucosides, two galactosides, a mannoside and one glucosamine building block. Simultaneously, Beau et al. reported a closely related procedure in which per-silylated glucosides were functionalized with a C-4, C-6 benzylidene acetal and a C-3 benzyl ether using benzaldehyde and Cu(OTf)2 catalysis (Scheme 2) [6]. They also demonstrated the possibility to extend the one-pot reaction sequence with an acylation step or a reductive opening of the benzylidene acetal.
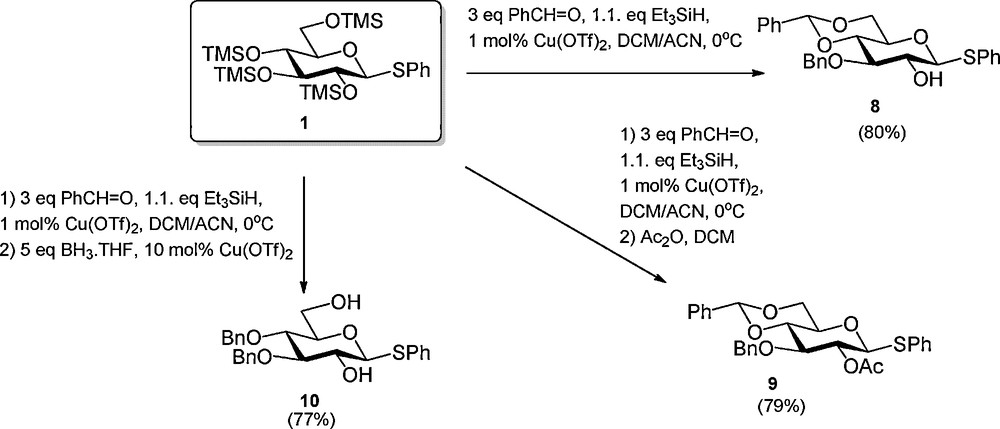
Cu(OTf)2 catalyzed one-pot regioslective protection of glucosides.
Stannyl ethers and dialkylstannylene acetals have found wide applications in the regioselective protection of carbohydrates [7] ever since their introduction in 1974 [8]. Onomura et al. recently described the use of a catalytic amount of dimethyltin dichloride (Me2SnCl2) for the regioselective protection of various monosaccharides [9]. The regioselectivity in the Me2SnCl2 benzoylation was shown to depend on the relative stereochemistry of the hydroxyl functions present. A fully protected α-O-methyl glucopyranoside was obtained as depicted in Scheme 3. Benzoylation of glucoside 11 provided the C-2 acylated product in 82% yield and the subsequent tosylation occurred selectively at the C-6 hydroxyl to give the diol product. Next a tert-butyl carbonate was introduced at the C3-OH, after which phosphorylation of the remaining alcohol provided the fully functionalized glucoside 12.
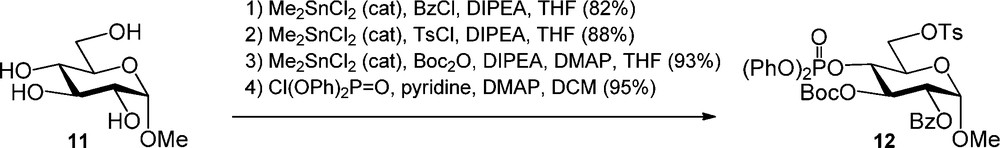
Dimethyltin dichloride catalyzed regioslective protection of glucose.
2 Protecting groups in the stereoselective construction of glycosidic bonds
Although the primary purpose of a protecting group is to prevent a given hydroxyl from reacting, it is now well established that the nature of the protecting group has a major effect on the reactivity [10,11] of a glycosyl building block and the stereoselectivity [12] and yield of a glycosylation reaction in which it partakes. This is best demonstrated by the a C-2 acyl protecting group in a donor glycoside [13], which not only deactivates this donor species as compared to its C-2 ether counterpart, but also reliably provides anchimeric assistance in the glycosylation process to provide 1,2-trans-glycosidic bonds (Scheme 4A). It is now becoming more and more clear that protecting groups at other positions than the C2-OH can also have a powerful stereodirecting effect. For example, Kim et al. recently demonstrated that the installment of an acetyl function on the C-3 hydroxyl of a mannosyl donor leads to the stereoselective formation of α-mannosides (Scheme 4B) through neighboring group participation [12b].
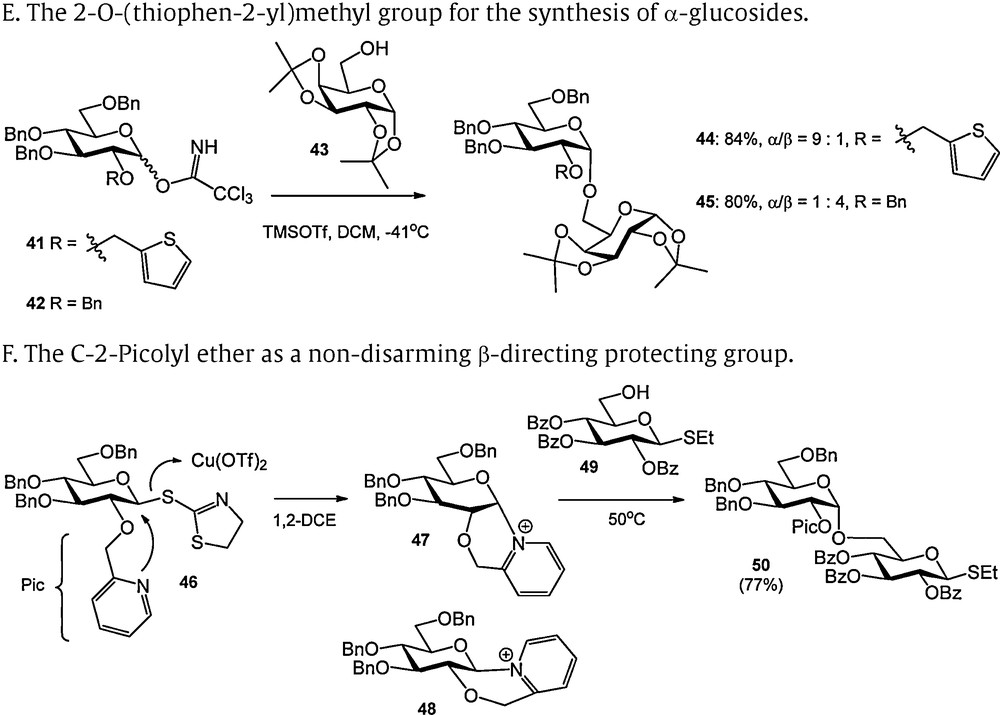
Protecting groups providing anchimeric assistance during glycosylation. A. Classical neighboring group participation by an acyl at C-2. B. Neighboring group participation by an acetyl group at C-3 in a mannoside. C. Chiral auxiliaries in the stereoslective formation of α-glucosidic linkages. D. Oxathiane donors for the synthesis of α-glucosides. E. The 2-O-(thiophen-2-yl)methyl group for the synthesis of α-glucosides. F. The C-2-Picolyl ether as a non-disarming β-directing protecting group.
The stereoselective formation of 1,2-cis glycosidic bonds is a long standing challenge in carbohydrate synthesis [14]. In 2005, the group of Boons developed two C-2 OH protecting groups that were capable of promoting the formation of 1,2-cis glycosidic bonds by neigbouring group participation [15]. As depicted in Scheme 4C, the (1S)-phenyl-2-(phenylsulfanyl)ethyl ether can be introduced at the C2 hydroxyl of glucoside 20 with (1S)-phenyl-2-(phenylsulfanyl)ethyl acetate 21 in the presence of BF3·OEt2. The configuration of the chiral center of the newly introduced ether is retained in this reaction because of the intermediate formation of an episulfonium ion, which is displaced at the benzylic position. Using standard protecting group manipulations, glucoside 22 was transformed into trichloroacetimidate 23. This donor could be (pre)-activated with TMSOTf to provide a meta-stable sulfonium ion 24a, having a trans-decalin structure. In this constellation, the phenyl substituent of the C-2 chiral auxiliary occupies an equatorial position. The alternative cis-decalin system 24b is not formed because this would place the phenyl group in an unfavorable axial orientation. Nucleophilic displacement of the intermediate sulfonium ion 24a provides the 1,2-cis product. The influence of the C2-chiral auxiliary was compared to the effects of an external sulfide, a non-chiral internal sulfide and the effect of the same auxiliary of opposite chirality. As can be seen in Scheme 4C, the best stereoselectivity was obtained with the (1S)-phenyl-2-(phenylsulfanyl)ethyl donor 23. Along the same line, the Boons laboratory developed the ethoxycarbonylbenzyl ether (as in 29) for the stereoselective construction of α-glucosyl and α-galactosyl linkages. It should be noted that the best selectivities were obtained with electron withdrawing protecting groups on the C-3 OH [16].
To circumvent the extra synthetic effort that is required to introduce the Boons auxiliary in a suitable donor, Turnbull reported oxathiane type donors such as 37 as depicted in Scheme 4D [17]. This type of donor can be activated by transformation into the corresponding sulfoxide 38 which can then be treated with triflic anhydride to provide a glycosylating species. Because the activated leaving group remains attached to the molecule upon glycosylation and thus ends up in the final product, the resulting sulfenyl triflate was transformed into arylsulfonium ion 39 by treatment with trimethoxybenzene. This species can be displaced with the acceptor alcohol. Because of the stability of the intermediate trimethylphenyl sulfonium ion, relatively high temperatures are required for this substitution and best results were obtained with primary alcohols. Notably the stereoselectivity of the glycosylations did not depend on the nature of the other protecting groups on the donor glycoside.
The 2-O-(thiophen-2-yl)methyl group was introduced by Fairbanks to provide a similar kind of anchimeric assistance as Boons’ phenyl-2-(phenylsulfanyl)ethyl ether (Scheme 4E) [18]. Good to excellent α-selectivities were reported for otherwise benzylated donors. No conditions were reported for the removal of the 2-O-(thiophen-2-yl)methyl group.
The trans-directing effect of the 2-picolyl ether (Pic) described by Demchenko et al. contrasts the α-directing effect of the sulfur-based participating groups described above (Scheme 4F) [19]. The C-2 picolyl ether was introduced as a “non-disarming” alternative for participating C-2 acyl functions. It was demonstrated that C-2 picolyl S-thiazolinyl donor 46 was transformed into a mixture of two bicyclic products, in which the α-oriented pyridinium ion 47 prevailed (47:48 = 20:1 at room temperature, 47:48 = 5:1 at 50 °C). The predominant formation of the 1,2-cis bicycle obviously differs from the generation of the β-sulfonium ions described above. Possibly, active participation of the picolyl ether in the expulsion of the S-thiazolyl under the mild activation conditions (Cu(OTf)2) is at the basis of this contrasting behavior. The α-pyridinium intermediate could be displaced by a glycosyl nucleophile at elevated temperature (50 °C) to provide the 1,2-trans products. The β-pyridinium ion proved to be inert under these conditions and was isolated after the reaction.
Besides the conceptually novel participating groups described above, several new acyl type protecting functions have recently been reported. For example, the 4-acetoxy-2,2-dimethylbutanoate was introduced as a pivaloyl analogue, that can be removed under relatively mild conditions (Scheme 5A) [20]. The 3-(2′-benzyloxyphenyl)-3,3-dimethylpropanoate ester has been developed as a participating group that can be removed by catalytic hydrogenolysis in concert with regularly used benzyl ethers (Scheme 5B) [21]. Other examples of recently introduced acyl protecting groups that are cleaved via a relay mechanism include the 2-(azidomethyl)benzoyl group [22] (Scheme 5C), the analogous 2-(azidomethyl)phenylacetyl ester [23], and the 2-(allyloxy)phenylacetyl group [24] (Scheme 5D). The 2-(azidomethyl)benzoyl ester (AZMB) was used as an orthogonal participating group in the synthesis of a protected H-type II pentasaccharide 63 by Seeberger et al., who also demonstrated its compatibility with various commonly used hydroxyl and amine protecting groups. Iadonisi et al. introduced alkoxycarbonates as participating functionalities to circumvent orthoester formation, which is a common side reaction when C-2-O-acyl protected donors are used in combination with mild activating conditions (Scheme 5E) [25]. We have reported on the methylsulfonylethoxycarbonyl (Msc) group as an orthogonal protecting group for hydroxyl functions in oligosaccharide synthesis that provides anchimeric assistance and excludes orthoester formation, when placed on the C2-OH of glycosyl donors (Scheme 5F) [26]. The group of Yamago showed that dialkyphosphate esters at the C-2 position are stereodirecting protecting groups for the synthesis of 1,2-trans-glycosides (Scheme 5G) [27]. In a pre-activation based glycosylation strategy [10d,e] thiophenyl glucoside 74 was activated using BSP/Tf2O at −60 °C, after which S-phenyl glucosamine synthon 75 was added. The thiodisaccharide 76 was obtained in good yield with excellent stereoselectivity.
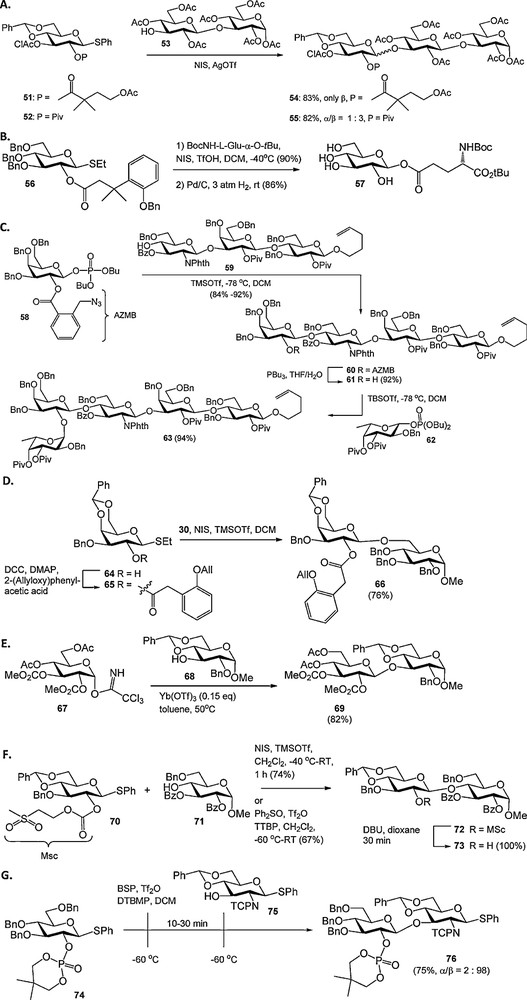
Novel participating acyl groups.
Protecting group size can have a major effect on the stereochemical outcome of a glycosylation reaction. For example, the bulky C-6 trityl ether has been shown to enhance the α-selectivity of glucosylations presumably by steric shielding of the β-face [28]. Crich et al. have reported on steric buttressing of large protecting groups at the C-3 hydroxyl in 2-O-benzyl-4,6-benzylidene mannosyl donors, which typically react in a highly β-selective fashion [29]. Placement of a large tert-butyldimethylsilyl (TBDMS) group at the C-3 hydroxyl in 78 caused erosion of the β-selectivity observed for its C-3 benzylated counterpart 77, because the large silyl group in 78 pushes the C-2 benzyl group towards the anomeric center of the mannosyl donor thereby obstructing nucleophilic attack on the β-face of the molecule (Scheme 6A). To overcome the poor selectivities of mannosyl donors with bulky C-3 substituents, Crich et al. introduced various propargyl ethers as minimally intrusive hydroxyl protecting groups. Firstly, the use of an unsubstituted propargyl ether was reported, which efficiently provided β-selective mannosylation reactions as exemplified in de coupling between 82 and 83 (Scheme 6A). Because the removal of the propargyl ether required a two-step sequence, namely base-induced isomerisation followed by oxidative cleavage of the intermediate allene ether by catalytic OsO4, substituted propargyl ethers were developed next (Scheme 6A). The 1-naphthylpropargyl ether can be cleaved in a single step using DDQ in wet DCM, but proved to be incompatible with the commonly used sulfonium activator systems BSP/Tf2O and Ph2SO/Tf2O. Furthermore when placed at the C-2 hydroxyl the 1-naphtylpropargyl group engaged in nucleophilic attack of the activated anomeric center. Therefore the 4-trifluoromethylbenzyl propargyl group was introduced. This ether was shown to be sterically minimally demanding, compatible with electrophilic activator systems and susceptible to cleavage in a single step using lithium naphthalenide (88 to 89, Scheme 6A).
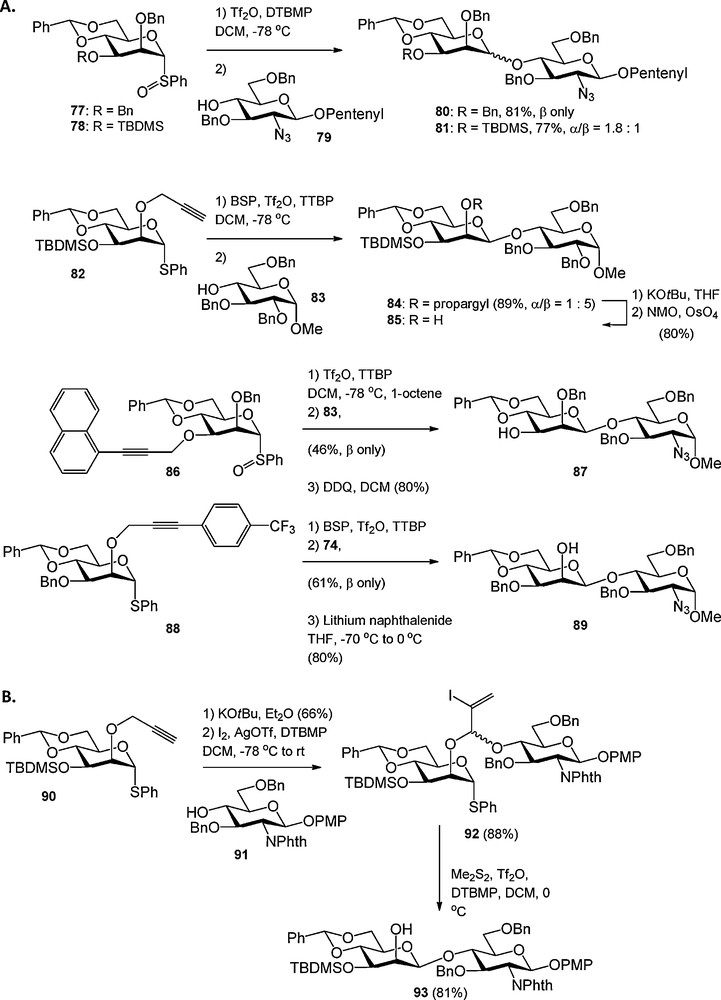
Propargyl ethers in carbohydrate chemistry.
The C-2 propargyl ether was exploited by Fairbanks et al. in an intramolecular aglycon delivery (IAD) strategy towards β-mannosides [30]. As depicted in Scheme 6B, the propargyl ether in 90 was isomerised to the allenyl ether, which provided the mixed acetal 92 upon treatment with a glycosyl alcohol (91) and iodine/AgOTf. Dimethyldisulfide-Tf2O [31] mediated intramolecular glycosylation led to the completely stereoselective formation of disaccharide 93.
Seeberger et al. described another solution to overcome the steric buttressing of the large TBDMS ether in 2-O-benzyl-4,6-O-benzylidene mannosyl donors described above. They showed that the tri-iso-propylsilyloxymethyl (Tom), in which the oxymethylene moiety moves the bulky silyl group away from the mannosyl core, could be used to install the β-mannosidic linkage [32]. The C-3-O-Tom mannosyl donor 96 was used in the automated solid phase synthesis of β-mannosides (e.g. 98, Scheme 7A). Our laboratory developed the methylsulfonylethoxymethyl (Msem) group as a new hydroxyl protecting group in oligosaccharide synthesis [33]. The Msem group is sterically unbiased and could be used for the synthesis of a 1,3-O-mannotrioside (103, Scheme 7B).
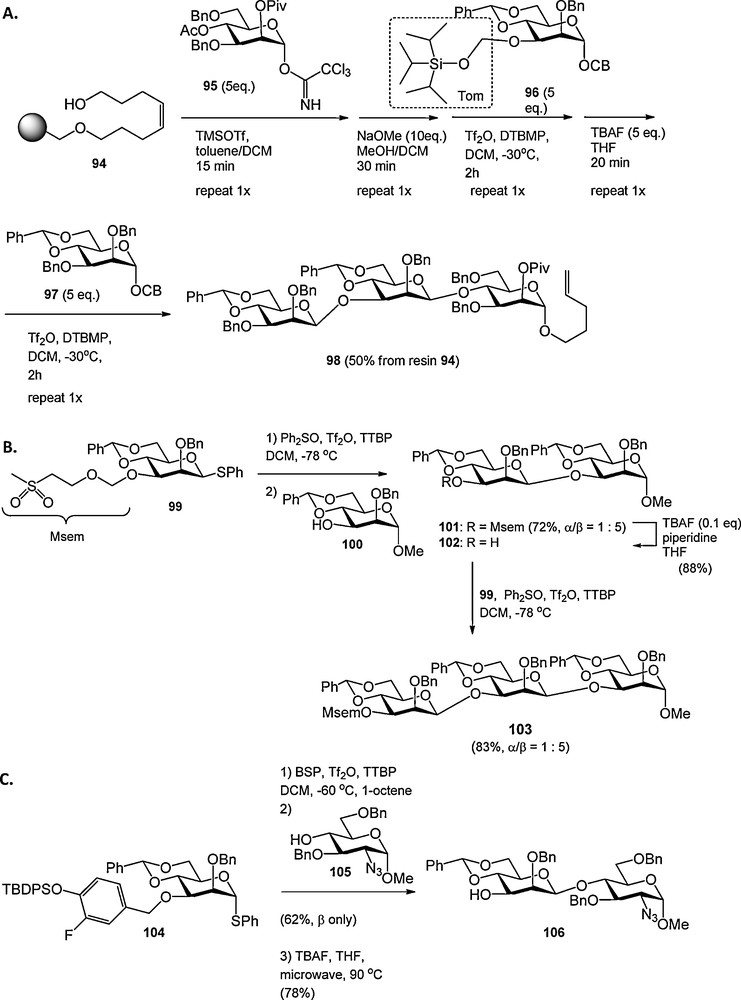
Fluorine labile, sterically minimally intrusive protecting groups in the construction of β-mannosides.
The 4-(tert-butyldiphenylsiloxy)-3-fluorobenzyl group was developed as a fluorine labile benzyl ether, attuned to the synthesis of β-mannosides (as in 104, Scheme 7C) [34]. The fluorine in this p-siloxybenzyl type ether was introduced to enhance its stability under acidic and oxidative conditions. The 4-(tert-butyldiphenylsiloxy)-3-fluorobenzyl ether could be formed from the corresponding benzyl bromide, which was synthesized in three steps from commercially available 3-fluoro-4-hydroxyl benzoic acid and cleaved with tetrabutyl ammonium fluoride (TBAF) at elevated temperature (90 °C) under microwave irradiation. Lower temperatures only led to removal of the silyl group.
The 4,6-di-tert-butylsilylene (DTBS) group was introduced in carbohydrate chemistry by Nishimura et al. as a more acid stable alternative to the commonly used cyclic ketal and acetal functions, such as the isopropylidene and benzylidene groups [35]. Dinkelaar et al. employed this group for the protection of glucosamine synthons in the assembly of a set of hyaluronan oligosaccharides, where the benzylidene group proved to be insufficiently stable towards the slightly acidic coupling conditions used [36]. Besides its acid stability, the 4,6-DTBS has attracted considerable attention because of the α-directing effect it has when mounted on a galactosyl donor [37]. Although the reasons for this stereodirecting effect are not completely clear yet, it has been hypothesized based on a crystal structure of a DTBS-protected N-phthaloyl thiogalactosamine that the near half chair conformation of the silylene group places one of the tert-butyl groups over the β-face of the galactosyl donor during glycosylation. Notably the α-directing effect is so strong that it can override neighboring group participation by C-2 acyl functions, such as the C-2-O-benzoyl, C-2-N-trichloroethylcarbonate (Troc), and C-2-N-phthaloyl (Phth) (Scheme 8).
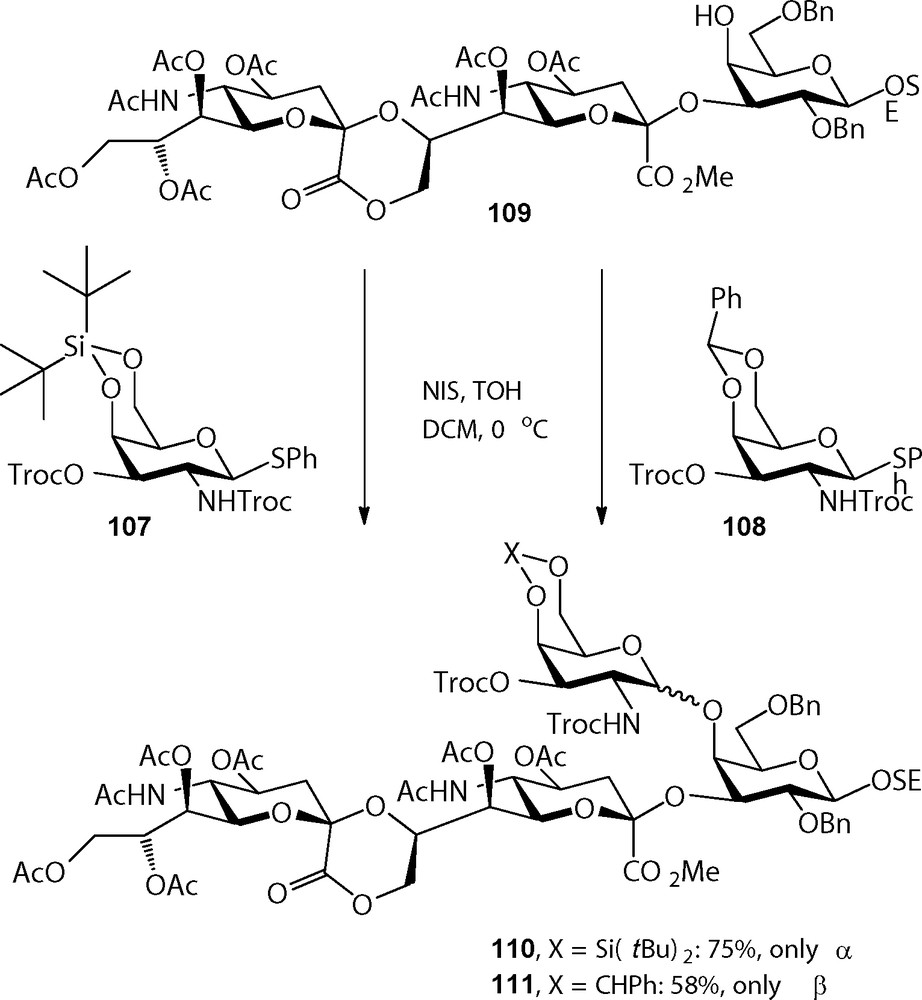
Stereoselective α-galactosylation using 4,6-silylidene galactosides.
The DTBS has also been applied in the stereoselective synthesis of β-arabinofuranosides [38]. Boons et al. proposed that the 3,5-DTBS group locks the arabinosyl oxacarbenium ion in the E3 conformation, which is attacked from the β-face in order to avoid eclipsing interaction on the α-face [39], to provide the 1,2-cis arabinosides (Scheme 9).
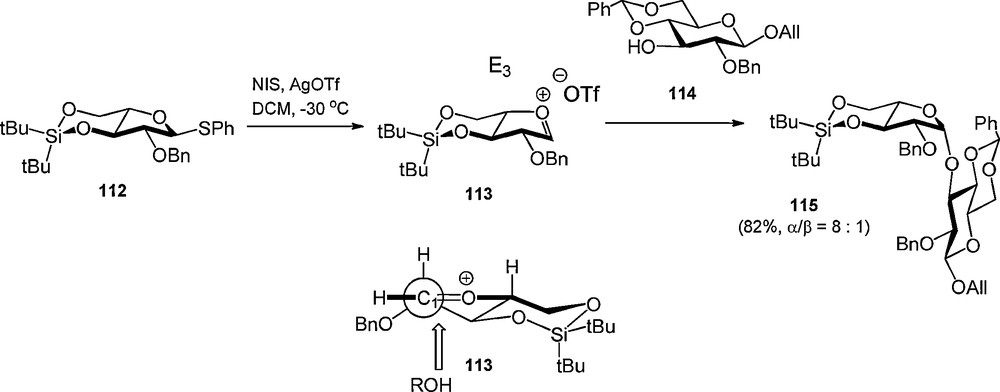
Stereoselective arabinofuranosylation using a 3,5-di-tert-butylsilylene group.
3 Protecting groups for the amino group in glycosamines
Several new protecting groups for the glucosamine nitrogen function have been reported recently. Schmidt et al. investigated several C2-symmetric N,N-diacyl groups, such as the dimethylmaloyl (DMM), diphenylmaloyl (DPM), dimethylglutaryl (DMG), diglycolyl (DG) and thiodiglycolyl (TDG) group (Scheme 10A) [40]. Of these the DG group proved to perform best in terms of ease of introduction, removal and compatabilty in glycosylation reactions. Yang and Yu introduced the N-dimethylphosphoryl (DMP) group for the protection of the glucosamine nitrogen (Scheme 10B). The DMP-group was used in the synthesis of several β-glucosamines, and shown to be stable to certain basic and acidic reaction conditions and could be readily removed using NaOH or hydrazine. Alternatively the N-DMP could be transformed into N-acyl derivatives using an acyl chloride in refluxing pyridine [41].
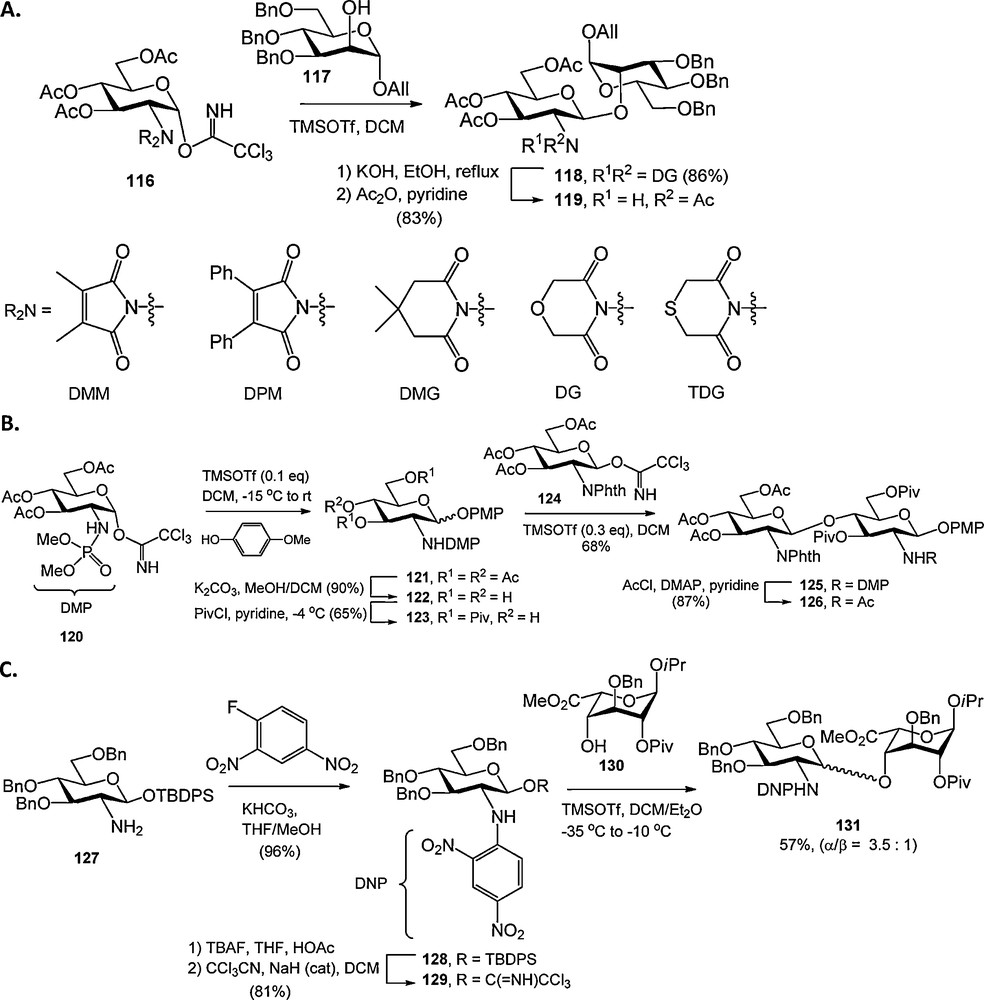
Novel glucosamine N-protecting groups.
Seeberger et al. explored the p-nitrobenzenesulfonamide (Nosyl, Nos) and dinitrophenyl (DNP) groups for the protection of the glucosamine nitrogen [42]. The synthesis of Nosyl protected glucosamine N-phenyltrifluoroacetimidate donors proved to be rather troublesome, and their glycosylations non-stereoselective. DNP protection of the glucosamine nitrogen proved to be more effective and condensations with an L-iduronic acid acceptor provided the α-linked disaccharide with moderate selectivity (Scheme 10C).
While there is a plethora of trans-directing nitrogen protecting groups, very few groups are available to mask the glycosyl amino function with a non-participating group [43]. In fact the azide group has almost exclusively been used for the installation of 1,2-cis glycosylamine linkages. In 2001 Kerns et al. introduced the oxazolidinone group for the protection of 2-aminoglycosides and it was shown that oxazolidinone protected glucosamine donors stereoselectively provided 1,2-cis linked products [44]. Mechanistic studies [45] revealed that the stereochemical outcome of condensations of these donors depends strongly on the nature of the activator and acceptor nucleophile used. Kerns et al. described that 2,3-oxazolidinone-N-acetyl protected glucosamine donor 132 can be activated with benzenesulfinylpiperidine (BSP) and triflic anhydride (Tf2O) in the presence of tri-tert-butylpyrimidine (TTBP), to mainly provide an α-anomeric triflate intermediate [45a]. Relatively reactive nucleophiles (such as 133) stereoselectively provided β-linked products, whereas the use of stererically hindered, less reactive nucleophiles (such as 134) led to the predominant formation of the α-products (Scheme 11A). These results were interpreted by assuming that the reactive nucleophiles can displace the α-triflate, but that less reactive nucleophiles can only substitute the more reactive β-triflate, or intermediate oxacarbenium ion. In subsequent studies the groups of Oscarson [45b,c], Ito [45d,e] and Ye [45f,g] showed that the stereochemical outcome of 2,3-oxazolidinone-N-acetyl and 2,3-oxazolidinone-N-benzyl protected glucosamine donors could be controlled by (Lewis)-acidity of the employed activator systems. Less acidic conditions mainly led to the isolation of β-linked products, where a more acidic milieu favored the formation of α-isomeric products. Convincing evidence has been forwarded that the glucosamine donors initially provide the β-linked products, which rapidly anomerise to the more stable α-isomers under acidic conditions through an endo-cyclic ring opening pathway (Scheme 11B).
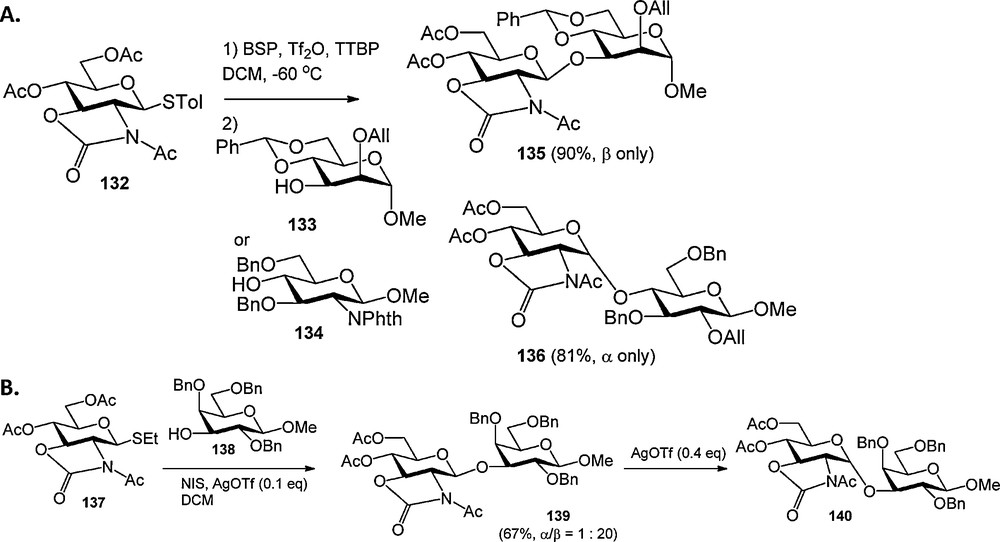
Stereoselective condensations of 2,3-oxazolidinone-N-acetyl protected glucosamine donors.
It is also of interest to note the beneficial effect of the 2,3-oxazolidinone-N group on the nucleophilicity of the C-4 hydroxyl in N-acetyl glucosamine acceptors [46]. The reason for this enhanced reactivity probably originates from the tied back nature of the oxazolidinone group, which makes the C-4 hydroxyl easier accessible and the disruption of potential H-bond networks.
The 4,5-oxazolidinone group has also been used as an N,O-protecting group for sialic acid in the synthesis of α-sialosides. Various groups have reported on the excellent stereoselectivity achieved with sialic acid donors bearing a 4,5-oxazolidinone group [47], culminating in various one-pot multi-step glycosylation procedures (Scheme 12) [47d,e].
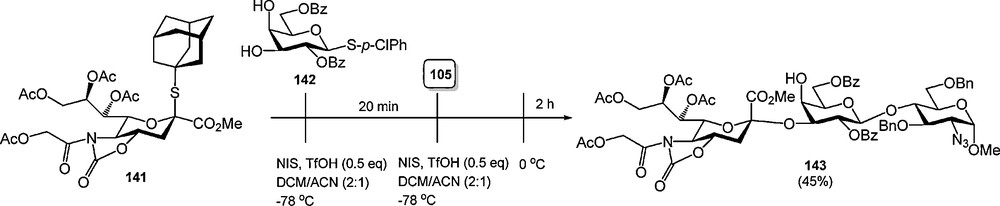
Stereoselective sialylations of 4,5-oxazolidinone protected N-acetyl sialic acids.
Removal of the oxazolidinone moiety can be accomplished using a variety of nucleophiles (NaOMe in MeOH, LiOH/LiCl in THF/H2O) but the selective removal of the oxazolidinone in N-acyl oxazolidinones has been shown to be difficult in many cases [47,48].
4 Protecting groups as visualization tags
Besides the primary role of masking a functional group on a carbohydrate, protecting groups can be used to introduce extra functionality in a carbohydrate building block. The UV-active 9-fluorenylmethoxycarbonyl (Fmoc) group has been extensively used as an amine protecting group in automated solid phase peptide chemistry to monitor the efficiency of the coupling steps [1d,49]. and has also found application as a hydroxyl protecting group in the (automated) synthesis of oligosaccharides [50]. Because the Fmoc is rather base labile when mounted on an alcohol, Pohl et al. set out to develop an alternative UV-active hydroxyl protecting group. They introduced the nitrophthalimidobutyric (NPB) ester, which can be introduced on a given hydroxyl function using the corresponding acid [51]. Cleavage of the NPB ester can be accomplished with hydrazine acetate in DMF at elevated temperature (50 °C) to provide the orange 3-nitrophthalhydrazide 146, which can be used in the colorimetric monitoring of reaction cycles. To illustrate its applicability Pohl et al. described the solid phase synthesis of resin bound glucosamine dimer 1 as displayed in Scheme 13. The aminomethylated SynPhase lantern support, functionalized with an allyl carbonate linker, was glycosylated with trichloroacetimidate donor 145 using a double glycosylation cycle. Cleavage of the NPB ester liberated the primary alcohol for further chain elongation and simultaneously allowed the colorimetric monitoring of the coupling efficiency, which was determined to be 98%. The second coupling/deprotection cycle proceeded in 96% yield. The dimer was not cleaved form the resin.
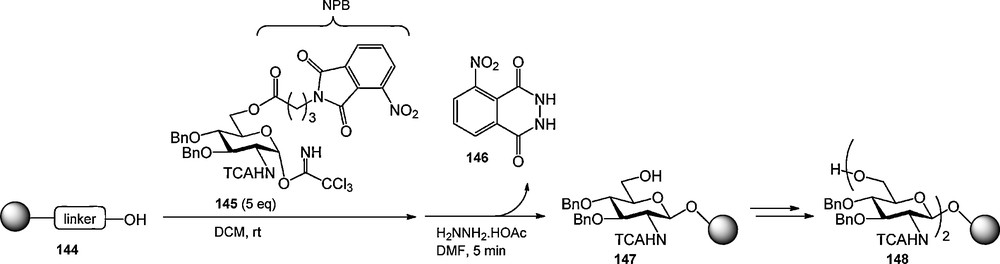
The NPB ester in the synthesis of resin-bound dimer 148.
The Seeberger laboratory introduced the UV-active 2-[dimethyl(2-naphtylmethyl)silyl]ethoxycarbonyl (NSEC) as a novel group to mask carbohydrate hydroxyl functions (Scheme 14) [52]. The NSEC group was shown to be compatible with various reactions commonly employed in carbohydrate synthesis and could be selectively cleaved with tetrabuylammonium fluoride (TBAF). The NSEC group was developed to allow UV-monitoring of glycosylation efficiency during automated synthesis but no such application has been reported yet.
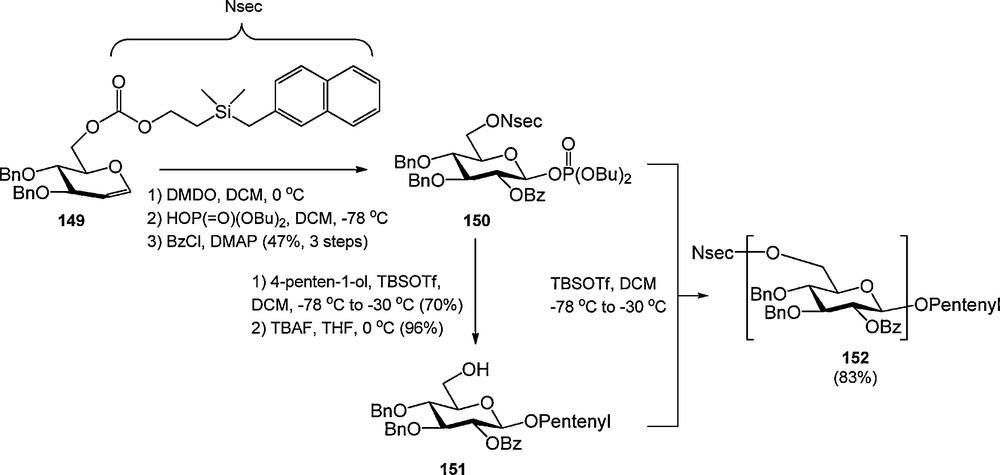
The NSEC group.
Several recent reports have described the use of azulene derived protecting groups (Scheme 15) [53]. Azulene is bicyclic hydrocarbon having a fused 7- and 5-membered ring and a 10-electron π-system, and an intense blue color. Lindhorst and Aumüller used guajazulene derived acid 154 for the protection of mannosyl alcohol 155 [53a]. After deprotection of the BDA-acetal, the mannoside was used in the synthesis of mixtures of acylated products. The blue color of the products allowed the visualization of the products during silica gel column chromatography. Timmer et al. introduced the azulen-1-yl-dicarbonyl (Az) group, which was used to protect a variety of carbohydrate alcohols [53c]. It was introduced from the corresponding acid chloride, and could be removed using catalytic NaOMe in methanol or diaminobenzene and acetic acid in refluxing ethanol. The latter deprotection conditions allowed the selective removal of the Az-ester in the presence of acetyl groups, providing the colored benzopyrazine 161 as a side product. The Az-group was shown to be compatible with glycosylation conditions involving trichloroacetimidate donors (catalytic TMSOTf), but NIS-TfOH mediated activation of an Az-protected thiophenyl galactoside led to thiophenylation of the Az-group. The color of the Az-group aided in the monitoring of reactions by TLC analysis and purification via column chromatography.
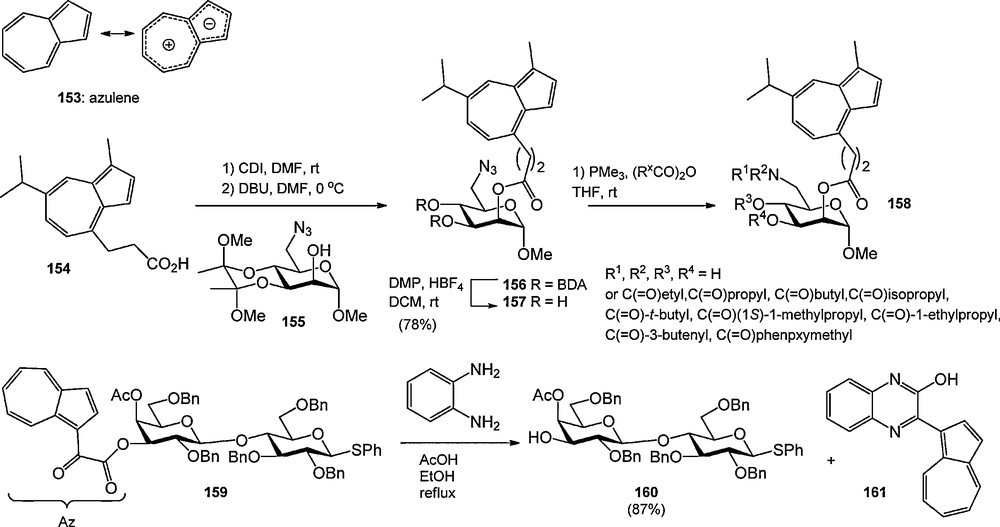
Azulene based protecting groups in carbohydrate chemistry.
5 Protecting groups as purification handles
Fluorous chemistry has been applied in various chemical areas, including catalysis, combinatorial and parallel synthesis, and selective tagging of biomolecules [54]. The properties of fluorous tags, being both hydrophobic and lipophobic, have been widely used in protecting group chemistry. Both “heavy” and “light” fluorous tags have been introduced as purification handles and their use has been extensively reviewed. “Heavy” fluorous groups are characterized by the presence of many fluorine atoms (39 or more) on multiple alkyl chains, often described as ponytails. The high fluorine content of these groups renders the molecules to which they are attached soluble in a fluourous solvent but insoluble in both organic and aqueous solvents. “Heavy” fluorous compounds can therefore be purified from non-fluorous compounds by simple liquid-liquid extractions. “Light” fluorous groups contain significantly less fluorine atoms, typically between 9 and 17, than their heavy counterparts. Because of the lower fluorine content, these molecules are often cheaper, easier available and much more soluble in organic solvents. Purification of “light” fluorous compounds can be effected by fluorous solid phase extraction (TSPE) techniques. Light fluorous versions of the most commonly used carbohydrate protecting groups have been described, including fluorous benzyl [55] trityl [56] allyl [57] and pentenyl [58] ethers, the fluorous benzylidene acetal [59], and various fluorous acyl [60] and silyl [61] based groups.
In analogy to oligosaccharide synthesis on solid or soluble polymeric supports, two strategies can be followed in the fluorous supported assembly of oligosaccharides. In a “donor-bound” strategy, the growing oligosaccharide chain is built up from the non-reducing end, with a donor glycoside bearing a fluorous protecting group/tag. The “acceptor-bound”strategy on the hand starts with a fluorous acceptor building block. Both strategies have been employed, but the latter has found most applications, because most side reactions in a glycosylation reaction take place on the donor glycoside. Liu et al. described the synthesis and application of fluorous glycosyl donor, in which the light fluorous tag was attached to the C-6 hydroxyl via a di-iso-propylsilyl ether [61c]. As depicted in Scheme 16, S-tolyl glucoside 162 was silylated with fluorous di-iso-propylsilane under the agency of triflic acid (TfOH) and subsequently converted into trichloroacetimidate 164. This donor was glycosidated with an excess of primary acceptor 162 to provide the disaccharide 165 in 93% yield. The S-tolyl disaccharide was transformed into a trichloroacetimidate to be coupled to reducing end glucoside 166 in the next step. After both glycosylation steps, FSPE was used to purify the products and recover the excess acceptor. Purification by silicagel chromatography was required in the transformation of the thioglycosides into the corresponding trichloroacetimidates. The fluorous di-iso-propylsilyl ether was cleaved at the end of the synthesis using 0.02 M HCl in MeOH/H2O.
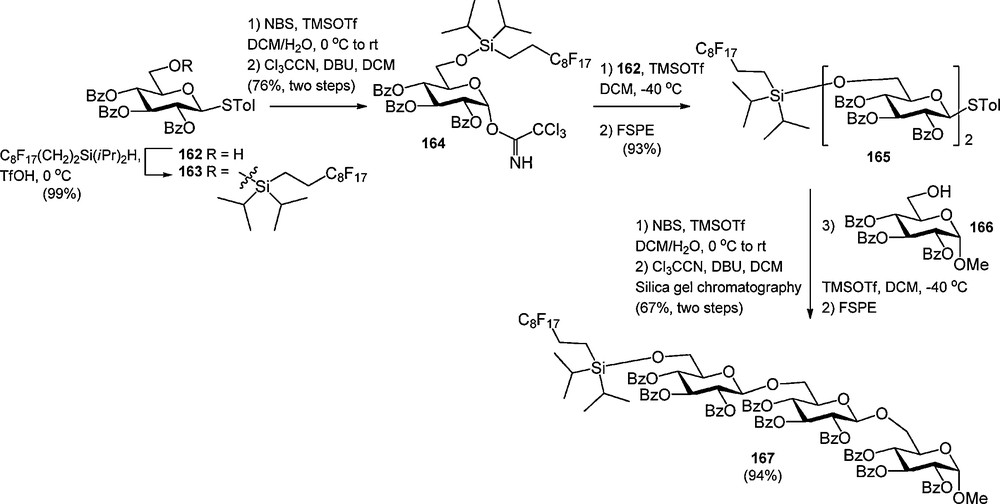
Use of a fluorous di-iso-propyl silyl glucosyl donor in the synthesis of a trisaccharide.
An example of an “acceptor-bound” oligosaccharide assembly strategy is depicted in Scheme 17. In 2007, Seeberger et al. introduced the fluorous version of the n-pentenyl group (169), which was employed in the assembly of a tetrasaccharide [58]. A silicon-glass microreactor was used for the optimization of the glycosylation reactions. It was described that β-glycosyl phosphate donor 168 could be employed at room temperature using reaction times ranging from 20 to 60 seconds. The first glycosylation had to be executed in a mixture of dichloromethane and trifluorotoluene (TFT) to keep the fluorous pentenyl alcohol 169 in solution. The lipophilic C-6 Fmoc protecting group was removed in the quenching step to facilitate purification by FSPE. After oligosaccharide assembly, the fluorous n-pentenyl group could be cleaved using N-bromosuccinimide, of transformed into different functional groups.
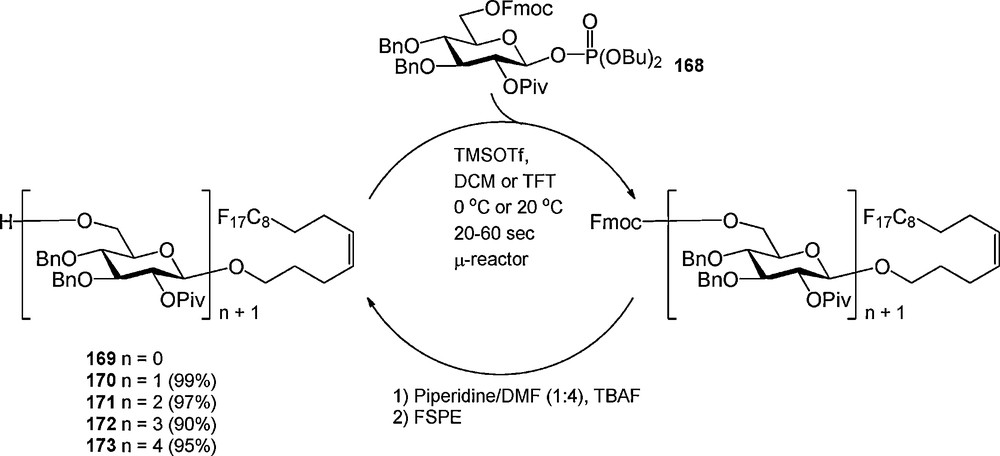
Fluorous oligosaccharide synthesis in a microreactor.
We recently introduced the fluorous version of the Msc-group [33]. It was found that an ethylene insulator, which is commonly used to spacer a fluorous tail from a functional group in a fluorous protecting group, made the F-Msc very base labile. Therefore the C8F17-moiety was removed from the sulfonyl group by a C3 spacer to provide the fluorous propylsulfonylethoxycarbonyl (FPsc) group. This sulfonyl carbonate was successfully employed in the synthesis of a trisaccharide as depicted in Scheme 18. First the FPsc was regioselectively introduced at the primary alcohol of glucoside 174. The resulting acceptor was coupled with levulinoyl protected thioglucoside 177 to provide the fluorous dimer 169 in 93% yield after FSPE. Deprotection of the Lev-ester then gave disaccharide 179 (81% after FSPE), which was elongated with glucosamine 180 to yield the trisaccharide 181 in 78% after FSPE.
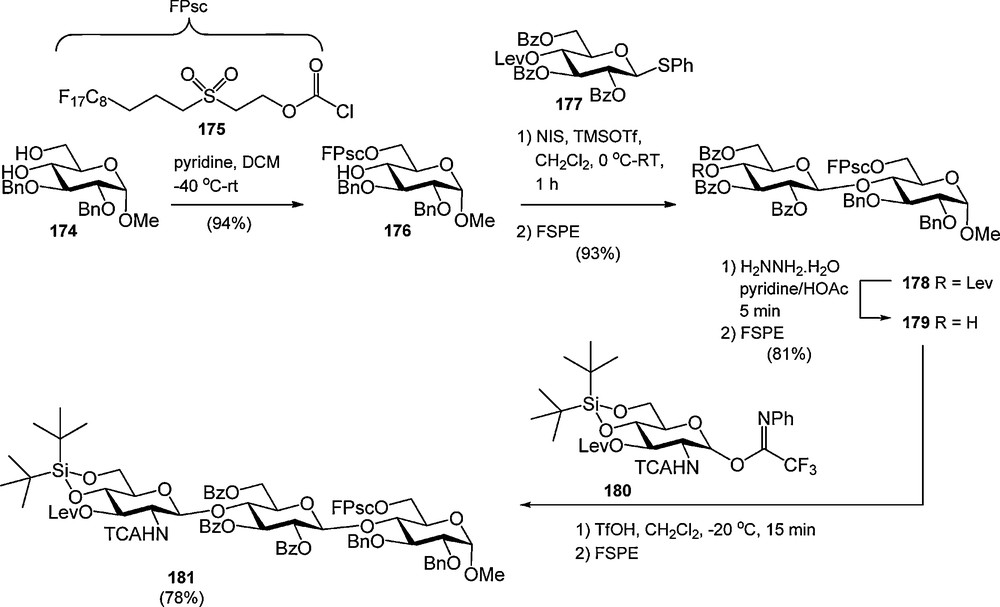
The fluorous propyl sulfonylethoxycarbonyl (FPsc) group in the synthesis of trimer 172.
Besides fluorous supports, large lipophilic moieties [62] and ionic liquids [63] have also been recently introduced for the construction of oligosaccharides.
6 Conclusion
Protecting groups take up a central role in carbohydrate chemistry and hold a key position in controlling the stereoselectivity of glycosylation reactions. Over the years, several new and ingenious protecting groups have been added to the broad palette of carbohydrate protecting groups to allow the stereoselective construction of both 1,2-cis and 1,2-trans linkages. New protecting groups, with tailor-made properties in terms of chemical stability and lability, allow ever more sophisticated glycosylation schemes to be developed, while colored or UV-active groups and purification tags continue to increase the efficiency of oligosaccharide assembly. It should be kept in mind however that a protecting group strategy which has been developed for one type of sugar, does not automatically translate well to other types of carbohydrates. Bridging this gap will probably require significant synthetic efforts in the future. Given the growing demand for ever more and complex, pure and well-defined oligosaccharides in all fields of glycoscience, it is anticipated that the development of new protecting groups and protection/deprotection schemes will continue to be a major theme in carbohydrate chemistry.
1 The 2-naphtylmethyl ether has found wide application in carbohydrate synthesis over the last decade. It has been used as a valuable alternative to the p-methoxylbenzyl ether, which is more acid labile than the 2-naphtylmethyl ether. Both ethers are readily cleaved under oxidative conditions (CAN or DDQ) or by catalytic hydrogenation.