

1 Introduction
Nanoparticles with a diameter of less than 10 nm have generated intense interest over the past decade due to their high potential applications in areas such as sensors [1–3], nanoscale electronics [4–6], catalysis [7–9], and optics [10]. The most important problems for the application of nanoparticles have been the oxidation reaction and the irreversible particle aggregation, which result in property changes over time and poor solubility in organic solvents. Self-assembly of organic monolayers on the nanoparticle surface can enhance the stability and solubility. Furthermore, recently developed synthetic methods of monolayer-protected metal nanoparticles offer the possibility of controlling both nanoparticle size and surface ligands in quantitative and qualitative manners [11].
Since the precise control over the chemical and structural composition of a monolayer surrounding a metal nanoparticle can dramatically affect its macroscopic properties and functions, functionalized monolayer-protected nanoparticles have found increased use in many recent applications including electrocatalysis [12], films [13], heavy metal detection [14], and chemical recognition [15,16]. This account focuses on the functionalization of monolayer-protected metal nanoparticles by means of organic reactions and application of these materials.
2 Synthesis of monolayer-protected metal nanoparticles
In 1981, Schmid et al. synthesized the ligand-stabilized gold nanoparticles, Au55(PPh3)12Cl6, for the first time [17]. However, the dependable and practical formation of more stable and isolable monolayer-protected metal nanoparticles was achieved less than a decade ago by Schiffrin et al. [18]. The alkanethiolate monolayers, which bind to the surface of a gold nanoparticle, prevent aggregation of the metal nanoparticles, stabilize them from harsh reaction conditions, and enhance their solubility in organic solvents. Subsequent investigations extend to other nanoparticle cores including Ag [19], Cu [20], Pd [21,22], Pt [23], and alloys [24–26] as well as various monolayers with different alkyl chain lengths and functional groups [11,27,28]. Other sulfur-containing organic compounds such as dialkyl disulfide [29,30], dialkyl sulfide [31–33], tetradentate thioether [34], methylthiirane [35], alkylthiosulfate [36], and xanthate [37] have been used to generate organic monolayer-protected metal nanoparticles. In addition, alkylamine [38], alkylisocyante [39], alkaneselenide [40], alkanetelluride [40], and phosphine [41] have been used for the synthesis of ligand-stabilized metal nanoparticles.
3 Synthesis of functionalized monolayer-protected metal nanoparticles
In the past few years, it has been shown that monolayer-protected nanoparticles can contain a wide range of functionalized tail-groups. These include acidic groups that can control flocculation properties and electroactive groups that can transfer these nanoparticles into redox-active nanoelectrodes [11]. The modification of nanoparticles by the incorporation of a functional group is of potential significance for the expansion of chemical and biological applications of these nanomaterials. There are two approaches to functionalizing the surface of monolayer-protected nanoparticles. One method is modifying the monolayer-protected nanoparticles by the ligand-replacement, which is described in detail in the following section. The other method is to synthesize the functionalized monolayer-protected nanoparticles directly from organic compounds with different functional groups (e.g. ω-functionalized alkanethiols [11,27,28], mixture of alkanethiols [42,43], unsymmetrical dialkyl disulfides [30], or sulfides [31]). The latter approach is found to be more useful for the synthesis of functionalized monolayer-protected nanoparticles with a higher loading of functionalized ligands.
4 Organic reactions of monolayer-protected metal nanoparticles
This section provides a survey of organic reactions, including ligand place-exchange reactions, used for the preparation of ω-functionalized monolayer-protected metal nanoparticles. Ligand place-exchange reactions paved a way for additional modification of ω-functional groups. A variety of organic reactions based on the chemistry of the reactive ω-functional groups are described.
4.1 Ligand place-exchange reactions
Nanoparticles with protecting monolayers composed of thiolate ligands can be functionalized by a partial ligand-replacement. In the exchange reaction, the incoming ligands replace the thiolate ligands on nanoparticles by an associative reaction and the displaced thiolate becomes a thiol. The rate of ligand-exchange depends on the chain length and/or steric bulk of the initial monolayers on nanoparticles. The longer the alkyl chain of monolayers, the less ligand-replacement occurs [44]. The ligand place-exchange at higher loading (>1:3) is inhibited due to the presence of steric repulsions [11]. In addition, bulky or small incoming ligands are difficult to replace the original ligands on the nanoparticle surface due to either kinetic (e.g. steric hinderance) or thermodynamic effects [11]. The rate and extent of ligand place-exchange reaction increase with increasing positive electronic charge on the Au nanoparticle core. By removing electrons from the core, the liability of the slightly more ionic Au-S bonds becomes further enhanced. When the gold core is charged to +2, up to 85% of the ligands can be replaced with incoming thiolate ligands [45].
Various functional groups ranging from simple organic functional groups to functional complexes are incorporated into the nanoparticles using ligand place-exchange reactions (Fig. 1). Among simple organic functional groups, the chemistry of ω-carboxylate group has drawn the most interest. Incorporation of carboxylic acid (COOH)-functionalized alkanethiols (e.g. 11-mercaptoundecanoic acid) results in the formation of amphiphilic nanoparticles. The nanoparticles are soluble in basic aqueous solution but aggregate in acidic solution by the formation of network via hydrogen-bonded carboxylic acids. The presence of the COOH group provides the controlled aggregation of particles through modulation of pH [46]. Similar pH-tuned nanoparticle networks show imparting biomimetic ion-gating recognition properties [47]. Metal ion chelating properties of ω-carboxylate-functionalized metal nanoparticles have been studied [48] and applied for heavy metal detection [14], nanoparticle film [49], and metal thin film [13]. ω-Pyridine-functionalized gold nanoparticles, synthesized by the ligand exchange reaction, can also be assembled on the solid substrates using a metal ion–pyridine complexation [50]. Nanoparticle films with metal ion linkers exhibit well-defined quantized double layer charging properties, electron hopping conductivity, and vapor sensing properties [51]. Monolayer-protected gold nanoparticles with ω-tetraalkylammonium ligands can interact with the DNA backbone via electrostatic interactions, which are highly efficient and completely inhibiting DNA transcription [52].

Ligand place-exchange reactions.
Various electroactive monolayer-protected metal nanoparticles such as ferrocene [53, 54], anthraquinone [12, 55], phenothiazine [56], galvinol [57], tetrathiafulvalene [58], nitrophenyl [59], and C60-functionalized metal nanoparticles [60,61] have been synthesized by ligand exchange reactions. Astruc’s ferrocene-terminated gold nanoparticles show interesting redox recognition of oxoanions [53]. Electrogenerated anthraquinone radical anions in the monolayers of nanoparticles catalyze the reduction of 1,1-dinitrocyclohexane [12]. Single-electron transfer properties of galvinol-functionalized nanoparticles are also being studied by Feldheim, et al. [57].
Binding a photoactive molecule to metal nanoparticles can enhance the photochemical activity of nanoparticles and provide potential applications for light-harvesting and optoelectronic applications [62]. Photoreactive ligands such as trans-stilbene and o-nitrobenzyl groups (6-thiohexyl-3-nitro-4-(4′-stilbenoxymethyl)-benzoate) are attached to nanoparticles and show preservation of normal solution-phase photoreactivity, but quench fluorescence [63]. Fluorophore (e.g. ethidium) bound gold nanoparticles are used as probes for biomolecular (e.g. DNA) labeling [64]. Fluorescent dansyl ligands [65], pyrene [66,67], photoswitching azobenzene derivatives [68], photoactive porphyrin [69] have also been incorporated into ligands on metal nanoparticles. Electrophoretically deposited C60-functionlaized gold nanoparticle assemblies on optically transparent electrodes show photoelectrochemical conversion of light energy [61].
Spin labels (e.g., nitroxide radical) are incorporated by a place-exchange reaction and these nanoparticles are used for EPR study [70]. ω-Dihydroquinidine-functionalized gold nanoparticles are used for the catalytic asymmetric dihydroxylation of olefins. The nanoparticles can be recycled with only a modest loss in efficiency after being isolated from the reaction mixture by gel permeation chromatography. [71] N-Methylimidazole-functionalized gold nanoparticles catalyze the cleavage of a carboxylic ester with more than an order of magnitude rate acceleration compared to acetyl-N-methylhistamine [72]. Other simple functional groups such as halide, nitrile, alkene, and sulfonate groups are incorporated by the ligand place-exchange reaction of monolayer-protected metal nanoparticles [73, 74].
4.2 Nucleophilic substitution reactions
In this preparation method for the functionalized monolayer-protected metal nanoparticles the nucleophile replaces the leaving group, which is bonded to the electrophilic carbon (Fig. 2). ω-Bromoalkanethiolate-functionalized gold nanoparticles exhibit a high SN2 reactivity with various alkyl amines, which is comparable to the reactivity of primary alkyl halide monomers (RCH2X) [75]. The reactivity largely depends on the steric bulkiness of the incoming nucleophile and the relative chain lengths of ω-bromo-alkanethiolate and surrounding alkanethiolate chains. The reaction retardation is resulted from the sub-merge of ω-bromo-functional groups and the use of bulky amines (e.g., tert-alkyl amine). ω-Hydroxyl-functionalized gold nanoparticles can also undergo nucleophilic substitution reactions with alkyl halide molecules, resulting in new ω-functionalized monolayer-protected nanoparticles [76].
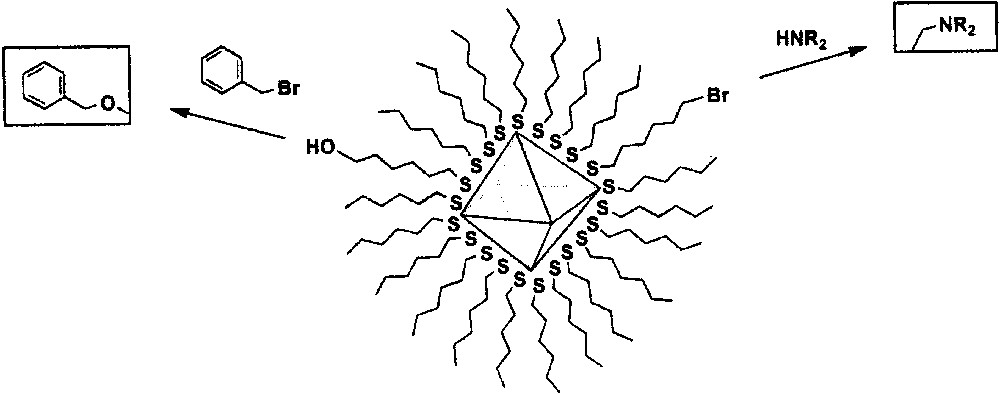
Nucleophilic substitution reactions.
4.3 Nucleophilic addition reactions
Nucleophilic addition of 4-aminothiophenoxide-protected gold nanoparticles to carbon-carbon double bonds of C60 generates C60-conjugated gold nanoparticles, as shown in Fig. 3 [77]. Furthermore, the addition reaction is applied for the layer-by-layer assembly of C60/nanoparticle conjugated films on the solid substrates (Fig. 4). This new nanostructure is expected to have interesting photonic and electronic properties.

Nucleophilic addition reactions.
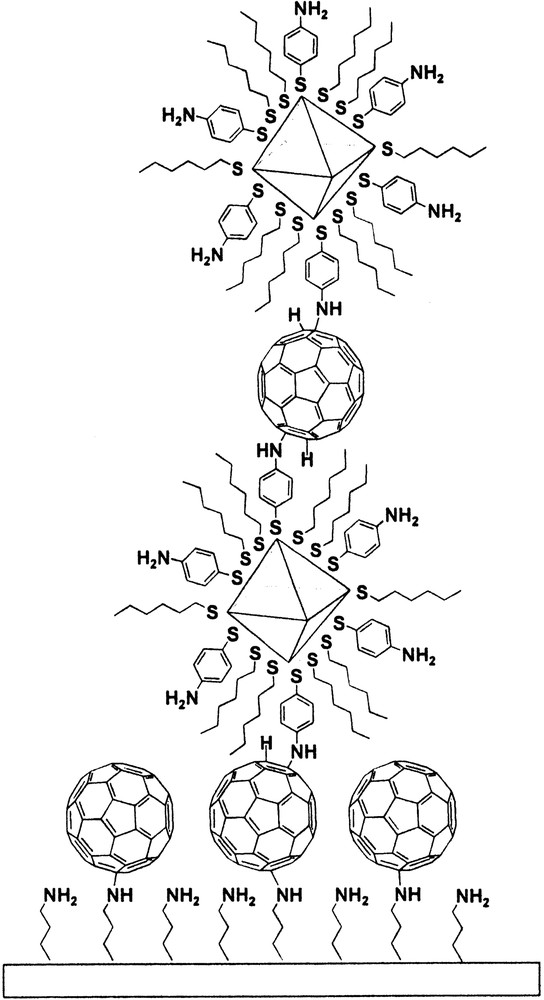
Layer-by-layer assembly of C60-conjugated nanoparticle films.
ω-Maleimido group-functionalized gold nanoparticles are used for the nucleophilic addition reaction of a sulfhydryl group attached to biological molecules such as a cysteine group on protein (Fig. 3). This nanoparticle can be used as a selective labeling agent for thiol-containing biomolecules including proteins, peptides, and oligonucleotides [2, 78].
4.4 Additions to carbonyls
Yu et al. prepared the ω-ketone-functionalized gold nanoparticles and immobilized them to self-assembled monolayers (SAMs) containing aminooxy groups on a gold surface (Fig. 5). Addition of amine moiety to carbonyl groups of nanoparticle is the driving force for this nanoparticle assembly. They also assembled gold nanoparticles protected with ω-aminooxy-functionalized alkanethiol onto ω-aldehyde-functionalized glass substrates [79].

Addition reactions to carbonyls.
ω-Aldehyde-functionalized gold nanoparticles can undergo addition reactions with amine-containing moieties to form a Schiff’s base or a stable covalent bond by reduction with sodium cyanoborohydride. Some protein molecules, including bovine serum albumin and lysines or some enzymes, may be labeled by organic reaction of ω-aldehyde-functionalized gold nanoparticles. ω-Amino-functionalized gold nanoparticles may be used to label carbohydrate moieties of glycoproteins, because they can react with aldehyde groups formed by subsequent oxidation of ribose cis diols (sugar) (Fig. 5) [80].
4.5 Substitutions at the carbonyls
Amide and ester coupling reactions of ω-functionalized monolayer-protected metal nanoparticles are used for the preparation of nanoparticles with various structural groups including peptides, nucleotides, sugars, spin labels, chromophores, fluorophores, electroactive groups, and ionophores (Fig. 6) [11,81]. This synthetic method offers a versatile way to functionalized nanoparticles, because safe and powerful activating agents are readily available and various target substituents can be easily obtained. This section is categorized into different types of coupling reactions based on the functional groups of incoming ligands and monolayers.
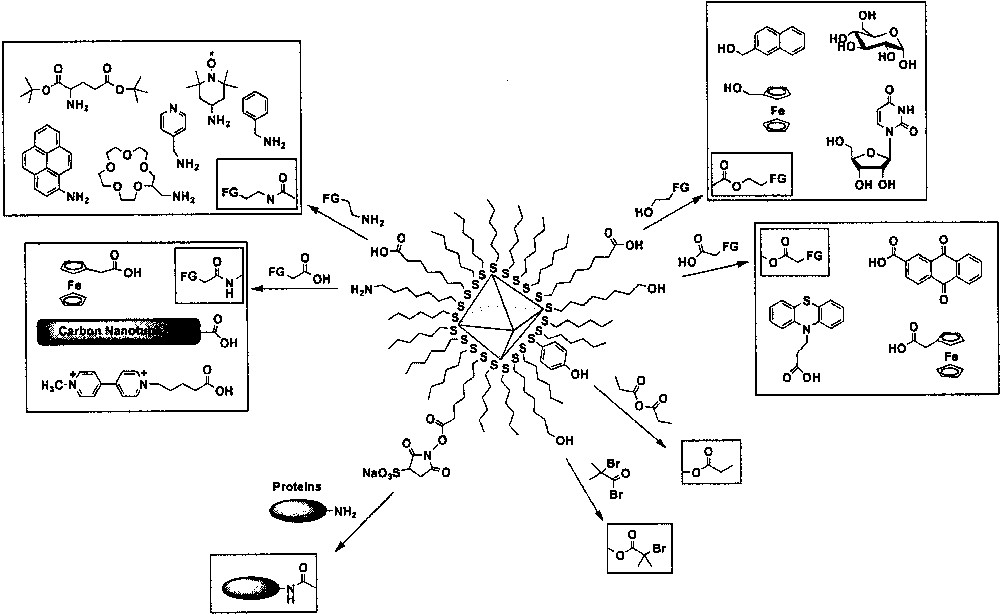
Substitution reactions at the carbonyls.
4.5.1 Carboxylic acid-alcohol-coupling reactions
ω-Carboxylic acid-functionalized gold nanoparticles are reacted with various organic compounds containing a hydroxyl group. For example, 2-nanpthaleneethanol, ferrocene methanol, α-d-glucose, and uridine are coupled to nanoparticles by the formation of ester linkage [81]. ω-Hydroxyl functionalized gold monolayer-protected nanoparticles also react with carboxylic acid-terminated compounds to generate new functionalized gold nanoparticles. The examples include the incorporation of phenothiazine and anthraquinone groups as electroactive labels [81]. ω-Hydroxyl-functionalized Pd nanoparticles also react with carboxylic acids, generating an ester linkage. Ferrocene group is incorporated into Pd nanoparticles for the synthesis of electroactive monolayer-protected Pd nanoparticles [82].
4.5.2 Carboxylic acid-amine-coupling reactions
ω-Carboxylic acid in the monolayers can react with compounds having amine moieties such as 4-amino-TEMPO, 4-(aminomethyl)pyridine, glutamic acid di-tert-butyl ester, 1-aminopyrene, 2-(aminomethyl)-15-crown-5, 5-(aminoacetamido)-fluorescein, and benzylamine [11, 81]. ω-Amino-terminated nanoparticles are used for the attachment of carbone nanotubes [83,84], ferrocene-carboxylic acids, and viologen-functionalized alkane carboxylic acids [85].
4.5.3 Carboxylic acid derivative-alcohol-coupling reactions
p-Mercaptophenol-protected gold nanoparticles are reacted with propionic anhydride, giving a propionate as a product of anhydride coupling reaction. However, only a partial esterification is observed due to steric constraints presented on the particle surface closely packed with p-mercaptophenol. [86] Living radical polymerization initiator can be incorporated into gold nanoparticles by reacting 11-mercaptoundecanol-functionalized gold nanoparticle with 2-bromoisobutyryl bromide. ω-hydroxyl groups can react violently with acyl halides and generate ester linkages [87].
4.5.4 Carboxylic acid derivative-amine-coupling reactions
Nanoparticles with reactive sulfo-N-hydroxy succinimide functional groups in the monolayers have a specific reactivity towards primary amines and may be covalently linked to any protein bearing an accessible primary amine [88,89].
4.6 Acid-base reactions
Acid-base chemistry of ω-functionalized metal nanoparticles has drawn quite an interest because electrostatic interactions play a crucial role in many chemical and biological processes [90,91]. Acid-base reactions of monolayer-protected metal nanoparticles have been mostly studied for molecular recognition [92,93] and nanoparticle film formation [94–96]. ω-Amino-functionalized metal nanoparticles have electrostatic interactions with acid containing organic and inorganic species. Carboxylic or sulfonic acid-functionalized nanoparticles react with base containing monomers and polymers (Fig. 7).
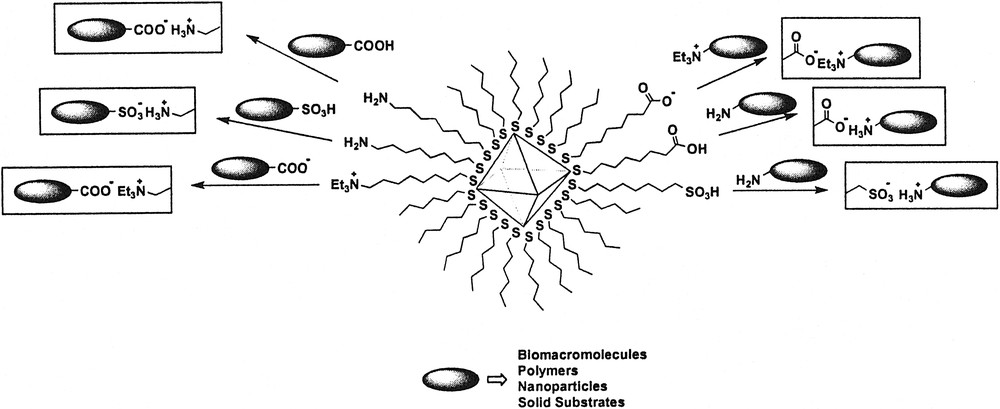
Acid–base reactions of monolayer-protected metal nanoparticles.
4.7 Polymerizations
Nanoparticles protected by a polymeric ligand have been employed to prepare nanoscale building blocks for functional materials. Polymeric stabilizers such as dendrimers [97], thiolated dendrons [98], thiolated polymers [99,100], and electrostatic polymer multilayers [101,102] have been used for the stabilization of a metal nanoparticle. Nanoparticles modified with polymer chains covalently bound to the surface using ‘grafting-from’ reactions are described (Fig. 8).
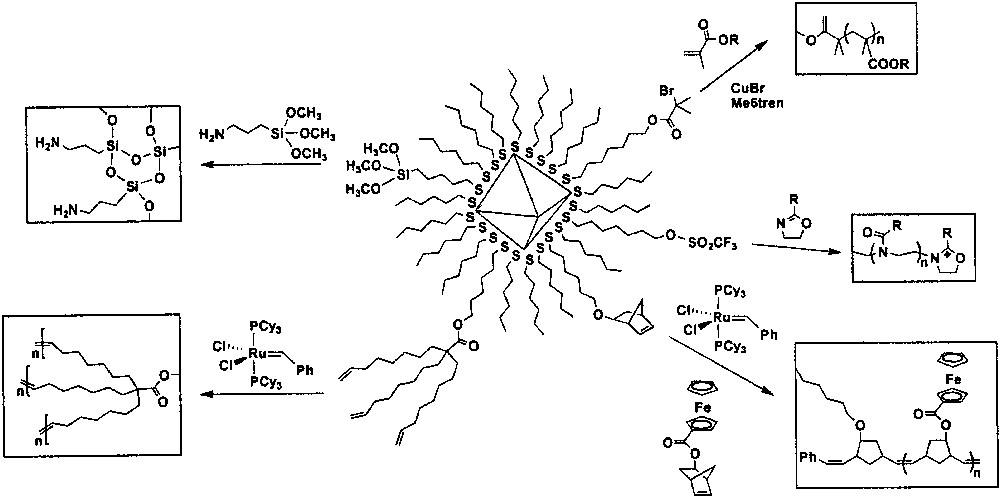
‘Grafting-from’ polymerizations on the monolayer-protected metal nanoparticles.
4.7.1 Radical polymerizations
Atom transfer radical (ATR) polymerization is one of the several methods for preparing polymer layers covalently bound to the surface of nanoparticles. ATR polymerization starting from initiator-modified gold nanoparticles is carried out with CuBr/Me6tren (Me6tren = tris(2-dimethylaminoethyl)amine) [103]. The same reaction is conducted using CuBr/1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane (Me4Cyclam) as a room-temperature catalyst [87]. These polymerization systems allow for the polymerization of n-butylacrylate [103] and methyl methacrylate [87,104] at room temperature.
4.7.2 Cationic polymerizations
ω-Trifluoromethanesulfonate-functionalized monolayer-protected metal nanoparticles are used to initiate the living cationic ring-opening polymerization reaction directly on nanoparticle surfaces. Dense polymer brushes are prepared in this one-pot reaction using 2-phenyl-2-oxazoline and N,N-di-n-octadecylamine as a monomer and a terminating agent, respectively [105].
4.7.3 Olefin metathesis polymerizations
ω-Norbornene group-immobilized gold nanoparticles allow a selective growing of polymer brushes off the surface of a nanoparticle with a surface-immobilized ring opening metathesis polymerization (ROMP) catalyst. Ferrocene group is also incorporated in the system using copolymerization of norbornene-containing nanoparticles and ferrocenyl-containing polymers using a ROMP catalyst [106]. Alkene-terminated tripodal ligands on gold nanoparticles can also undergo olefin metathesis polymerizaton, yielding polymer shell cross-linking. Nanoparticle etching of this polymer-nanoparticle composite produces spherical hollow polymer capsules [107].
4.7.4 Polymerizations of organosiloxanes
ω-siloxane-functionalized gold nanoparticles couple with functionalized siloxane monomers, forming polymeric siloxane shells surrounding the gold nanoparticle core [108]. These hybrid materials are an example of inorganic shell-protected nanoparticles. Polysiloxane chains are major components in glass and in a wide variety of mineralogical silicates.
4.8 Miscellaneous reactions
ω-Thiol-terminated monolayer-protected nanoparticles generated from a protected sulfhydryl-terminated monolayer can react with 2,2′-dipyridyl disulfide to form disulfide bonds. These disulfide bonds are used in a thiol-disulfide exchange reaction with free sulfhydryls in order to attach biomolecules such as thiol-containing DNA and cysteine-containing polypeptides [109].
5 Conclusions and future outlook
This account summarizes an increasing research field in nanosciences. The organic reactions of monolayer-protected metal nanoparticles have undoubtedly expanded the available diversity of the nanomaterials as functionalized chemical reagents. However, there is still a great demand for new and convenient techniques that allow the preparation of well-defined hybrid nanoparticles. More extensive and systematic studies are essential for further advancement in the nanoparticle-related research field. Building an extensive database with new reactions and conditions will be one thing. The sophisticated applications in, for example, chemical sensing and catalysis certainly will benefit from this progression. Construction of nanostructures using multi-step organic reactions of ligand-stabilized nanoparticles will be another. This would benefit the expansion of functional nanoscale building blocks for future applications.
Acknowledgements
Y.-S. Shon thanks supports from Research Corporation, Kentucky NSF ESPCoR, KSGC/NASA, and Western Kentucky University (Applied Research and Technology Programs).