

La dispersion de métaux plus ou moins radioactifs en cas d’accident nucléaire ou de conflit armé peut conduire, par inhalation ingestion ou blessure, à de graves intoxications, qui résultent de la toxicité chimique ou de la radioactivité.
Ces métaux (plutonium, américium, thorium, neptunium, uranium, cobalt) doivent être éliminés de l’organisme par l’administration de ligands non toxiques, qui permettent l’obtention in vivo d’un complexe stable et soluble, susceptible d’être éliminé par la barrière rénale ou hépatique (décorporation).
Alors que des ligands existent pour la décorporation du plutonium, de l’américium et du thorium, il n’existe pas à ce jour de ligands susceptibles d’éliminer l’uranium le neptunium et le cobalt d’une manière satisfaisante. En ce qui concerne l’uranium, des résultats encourageants ont été obtenus avec des ligands polydentés dérivant des hydroxypirydinones [1–4] et des hydroxy bisphosphonates [5,6].
Les bisphosphonates simples, tels que HEBP (hydroxy éthyle bisphosphonate), MDP (méthylène diphosphonate), MAMDP (N,N′-diméthylamino méthylène diphosphonate) (Fig. 1 ), sont capables de complexer le cobalt(III) [7] et l’uranium(VI) [8] ; les complexes formés A et B (Fig. 1) montrent que les bisphosphonates agissent comme des ligands bidentés. Toutefois, l’uranium(VI) peut également former avec les ligands bidentés des complexes de l’uranyle hexacoordinés dans le plan équatorial de la bipyramide hexagonale, comme dans le cas du tricarbonate d’uranyle, C, dont la structure a été déterminée par diffraction X [9].

Notre objectif est de synthétiser des ligands hexadentés par greffage de trois motifs, bidentés, bisphosphoniques comportant une chaîne de liaison à fonction carboxylique activée, 8, sur une molécule support aminée par couplage amidique (Fig. 2 ).
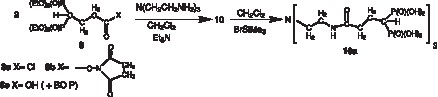
Le ligand obtenu devrait conduire avec l’uranium à un complexe de type ML (Fig. 3 ). En effet, il a été démontré, dans des études de complexation du fer en milieu très dilué (10–6 M) que les constantes de stabilité des complexes ML étaient largement supérieures à celles des complexes ML3, toutes choses étant égales par ailleurs [10]. De plus, les acides phosphoniques sont plus fortement ionisés que les acides carboxyliques ou que les catécholates à pH 7 ; leur atome d’oxygène donneur dur respecte la règle hard–soft pour établir une liaison de coordination avec l’atome d’uranium dur. Enfin, la formation d’un cycle à six chaînons est également favorable à la stabilité de l’ensemble (effet chélate).

Modèle présumé du complexe formé à partir du ligand 10a. Deux des « bras » du tripode n’ont pas été représentés.
La nature des supports aminés tripodes, 1–4, linéaires, 5, 6, ou cycliques, 7 (Fig. 4 ), ainsi que la longueur de la chaîne activée du bisphosphonate 8 (Fig. 2), ont été choisies pour être compatibles, sans contrainte stérique avec l’ion uranyle (Fig. 3). Ces choix ont été effectués en utilisant des modèles moléculaires Dreiding. L’atome d’uranium non disponible commercialement a été construit en utilisant les paramètres de diffraction X connus, et en particulier ceux du tricarbonate d’uranyle.
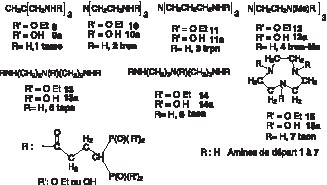
Les synthèses sont réalisées par addition de 3 équiv de diphosphonate 8 dans une solution d’un équivalent de triamine (1–7) dans CH2Cl2, en présence de quatre équivalents de Et3N sous agitation à 20 °C. Après 12 h d’agitation, le mélange est lavé à l’eau saturée en NaCl. La phase organique est décantée, séchée sur MgSO4 et évaporée sous pression réduite. Le résidu huileux (intermédiaires 9–15) est, soit purifié par chromatographie, si nécessaire, soit directement engagé dans l’étape suivante de désestérification. Le rendement moyen est de 75%.
Les ligands 9a–15a sont obtenus par dissolution des intermédiaires précédents dans CH2Cl2, auquel on ajoute 12 équiv de BrSiMe3. Après 4 à 7 jours de contact à 20 °C, le mélange homogène est évaporé à sec, sous pression réduite. Le résidu est repris par un excès de méthanol, puis évaporé à sec, cette opération étant répétée quatre fois, dans le but d’éliminer les produits secondaires silylés, très volatils. Le résidu de la dernière évaporation est, soit lyophilisé, pour obtenir les acides phosphoniques libres, soit traité par une solution méthanolique de soude, jusqu’à pH 7,5. Le sel de sodium formé est filtré et séché sous pression réduite.
Les polyamines de départ sont commerciales, à l’exception du tame (tris(aminométhyle)éthane) 1, du tren-Me (tris(2 aminoéthyle) méthyle amine) 4 et du trpn (tris(3-aminopropyle) amine) 3, qui ont été respectivement préparés par les méthodes décrites dans les références [11–13]. Le bisphosphonate 8c à fonction carboxylique libre a été décrit antérieurement [14]. Nous avons réalisé l’activation du groupe carboxylique par le carbonate de disuccinimide en présence de triéthylamine. Un essai d’activation par le BOP (réactif de Castro), en une étape, au moment du couplage amidique, a donné également un bon résultat, mais nécessite une purification laborieuse pour éliminer l’HMPT formé. La fonction carboxylique a également été activée sous forme de chlorure d’acide à partir du chlorure d’oxalyle.
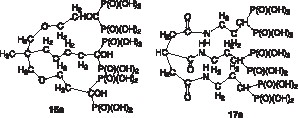
Les tripodes 16a [15] et 17a [16] (Fig. 5 ) ont été synthétisés, sur la base des critères formulés plus haut. L’objectif étant d’améliorer la stabilité du complexe formé avec UO2++ par rapport au HEBP décrit antérieurement. Cette approche s’est avérée concluante : en effet, les tests de décorporation sur 17a ont confirmé nos prévisions [16]. Dans 17a, la molécule support des trois groupements bisphosphoniques est l’acide carballylique, dont les trois fonctions CO2H ont été transformées en chlorure d’acide par le chlorure d’oxalyle. La réaction de couplage a été réalisée avec un bisphosphonate à fonction terminale NH2, analogue de 8.
Les paramètres RMN sont reportés dans le Tableau 1.
Paramètres RMN sur Bruker ARX 400 et Varian Inova 500
| N° | δ31P | δ 1H (ppm), J (Hz), δ13C (ppm), J (Hz) |
| 8b | 23,5 (CDCl3) | 1H:1,33 t. J 7 (12 H) ; 2,26–2,38 m (2 H) ; 2,42–2,56 tt. JHH 6,45 JHP 23,7 (1 H) ; 2,82 s. (4 H) ; 2,99 t. J 7,4 (2 H) ; 4,13–4,22 m (8 H). (CDCl3). |
| 13C: 16,8 ; 21,3 ; 26 ; 29,9 ; 35,9 t. JCP 133,8 ; 63,4 ; 168,5 ; 169,6. (CDCl3). | ||
| 10 | 24,3 (CDCl3) | 1H: 1,34 t. J 7,02 (36 H) ; 2,19–2,27 m (6 H) ; 2,54–2,67 m (15 H) ; 3,25 qu. J 5,39 (6 H) ; 4,12–4,19 m (24 H). (CDCl3). |
| 13C: 13,3 ; 21,8 ; 34,2 ; 35,3 t. JCP133 ; 37,24 ; 54,2 ; 62,8 ; 172,6. (CDCl3). | ||
| 10a | 22,3 dt. JPH 21,61 ; JPP 15,8 (D2O) (pH 4) | 1H: 1,84 t. –1,88 t. –1,94 t. JHH 6,59 JHP 21,97 (3 H) ; 1,95–2,1 m (6 H) ; 2,47 t. J 7,5 ; 3,31–3,34 m (6 H) ; 3,50–3,52 m (6 H). (D2O) (pH 4). |
| L'irradiation du signal 3,31–3,34 transforme le signal à 3,50–3,52 en singulet et inversement, soit δ (respectivement) : 3,33 et 3,51. | ||
| 11 | 21,4 (CDCl3) | 1H: 1,3 t. J7,8 (36 H) ; 1,7 m (6 H) ; 2,1–2,2 m (9 H) ; 2,5 t. J 7,8 ; 2,53–2,63 m (6 H) ; 3,22–3,26 m (6 H) ; 4,1–4,2 m (24 H). (CDCl3). |
| 13C : 16,4 ; 21,7 ; 25 ; 35,5 t. JCP 133 ; 36,8 ; 51 ; 62,7 ; 172,7. (CDCl3). | ||
| 11a | 20 (D2O pH 7,6). | 1H: 1,4–1,9 m (15 H) ; 2,2 t. J 7,8 (6 H) ; 2,8–3,2 m (12 H). (D2O pH 7,6). |
| 12 | 21,5 (CDCl3) | 1H : 1,27 J t. 6,8 (36 H) ; 2,12–2,2 m (6 H) ; 2,48 tt. JHP 24,4, JHH 5,86 (3 H) ; 2,56–2,69 m (12 H) + 2,57 d. J 9,76 (HMPT) ; 2,88 s, 2,95 s (9 H) ; 3,33 m (6 H) ; 4 m (24 H). |
| 13C : 16,4 ; 21,2 ; 32 ; 35,8 t. JCP 133 ; 36 ; 36,8 ; 51,4 ; 62,6–62,7 (4 pics) ; 172 (CDCl3). | ||
| 12a | 20,1 (D2O pH 14). | 1H: 1,6 tt. JHH 5,5 JHP 21,6 (3 H) ; 1,8–1,9 m (6 H) ; 2,5–2,6 m (12 H) ; 3 s. (9 H) ; 3,3–3,5 m (6 H). (D2O pH 14). |
| 13 | 24,4 (CDCl3) | 1H: 1,27 t. J 7,05 (36 H) ; 2,2–2,39 m (9 H) ; 2,61 t. J 6.65 (6 H) ; 2,8 m (4 H) ; 3,4 m (4 H) ; 4,13–4,23 m (24 H). (CDCl3). |
| 13a | 3s. 22,55 ; 22,60 ; 22,62 ; (1:1:1) (D2O pH 14). | 1H: 1,9–2,04 m. (9 H) ; 2,4–2,6m (6 H) ; 3,1–3,4 m. (8 H). (D2O, pH 7). |
| 15 | 21,5 (CDCl3) | 1H: 1,33 t. J 6,84 (36 H) ; 2,16–2,25 m (6 H) ; 2,56 t. J 6,84 ; 2,61 t. J 6,84 ; 2,65 t. J 6,8 (9 H)* ; 3,4 s large ; 3,6 s large (12 H) ; 4.14–4,19 m (24 H). (CDCl3). |
| 13C: 16,92 ; 21,73 ; 35,73 t. JCP 133,8 ; 36 t. JCP 133,5 ; 35,9 t. JCP 134,4 (**) ; 49 ; 51,4 ; 173,7. (CDCl3). | ||
| * triplet de triplet de H–C(P)(P) + triplet de CH2C(O). | ||
| ** C de H–C(P)(P), 3 triplets. | ||
| 15a | 23,7 (D2O, pH 14) | 1H: 1,47 * t. JHP 22,3 (3 H) ; 1,8–1,95 m (6 H) ; 2,5–2,82 m (6 H) ; 3,41–3,68 m (12 H). |
| * déplacé à 2,2 à pH 1 (D2O). |
Nous envisageons de poursuivre ce travail, avec la même stratégie, sur d’autres supports polyaminés ou polycarboxyliques tels que le cis,cis-triaminocyclohexane ou l’acide de Kemp. En effet, la stéréochimie de ces composés permettrait d’exploiter le concept de préorganisation et ainsi d’augmenter encore la stabilité des complexes formés in vivo.