

1 Introduction
During fly ash vitrification processes, heavy metals volatilise and are recovered into chlorinate salts, called secondary ashes [1]. The secondary ashes contain large amounts of chlorinated salts rich in highly soluble and volatile heavy metal chlorides, like lead and cadmium chloride. This is why the treatment of these toxic compounds must involve, in a first step, the conversion of the heavy metals into a thermally stable form [2]. Several processes have been proposed for this conversion. One involves dissolving chlorides in water followed by a precipitate of hydroxides in alkaline medium. The drawback is the handling of brine and energetic cost for eliminating water [3]. Ikeda et al. [4] have studied the thermal conversion reactions of metal chlorides into oxides with boric acid to develop a method for vitrification of radioactive molten salt wastes. But the melting occurred at 1100 °C and volatilisation still occurred at this temperature. We proposed an alternative way using a solid-state conversion of chlorides into phosphates, using ammonium hydrogenphosphate [2]. The reaction leads to the recovery of chlorides as ammonium chloride which sublimates around 300 °C [4], and heavy metals are transformed into non-volatile phosphates that can be vitrified at 900 °C. Tin chlorides are difficult to convert because they are much more volatile than lead and cadmium. Furthermore, Sn2+ can oxidise into Sn4+, which has a low solubility in glasses. It has been reported [5] that stannous chloride (SnCl2) reacts with ammonium dihydrogenphosphates to form glasses in the SnO–SnCl2–P2O5 system. The conversion of tin chlorides into phosphate using an ammonium phosphate process is efficient [6]. Tin is known to decrease glass transition temperatures of phosphate glasses [7]. However, very little is known about the structure of tin phosphate glasses. Holland et al. reported a NMR study of x SnO–(100 – x) P2O5 glasses [8], but did not considered the presence of Sn(IV), since its quantity was expected to be low. Contrary to Holland et al. [8], we paid a special attention to the effect of the redox ratio R = Sn2+/Sntotal, on the structure and properties of tin phosphate glasses, because oxidation of Sn2+ into Sn4+ is expected during vitrification of fly ashes. In order to investigate this effect on the structure of phosphate glasses, a series of x SnO–(100 – x) P2O5 glasses was synthesised and characterised with 31P and 119Sn NMR.
2 Experimental procedures
x SnO–(100 – x) P2O5 glasses were prepared from batches of reagent-grade SnO (Aldrich) and (NH4)2HPO4 (Fisher). Each batch was calcined in an alumina crucible at 200 °C to drive off NH3 and H2O and was then melted in a carbon crucible at 1000 °C for 2 h under argon.
The glasses were analysed by inductively coupled plasma atomic emission spectrometer (ICP–AES). The [Sn2+] concentration was analysed by polarography. The redox R is then calculated as R = Sn2+/Sntotal. The redox was also checked with tin Mössbauer on some glass compositions, and found in good agreement with solution measurements.
The glass transition temperature (Tg) was determined using a differential scanning calorimeter. The samples were heated at 5 K min–1 up to 450 °C. The reported Tg values are reproducible within ±5 K.
The 31P MAS-NMR spectra were recorded on a Bruker ASX100 spectrometer operating at 2.34 T, with a 1.5-μs pulse (45°), and a 60-s delay between each accumulation to enable full relaxation. A 7-mm MAS probe was used. The MAS–NMR spectra were decomposed with the DM-FIT software [9]. Gaussian lines were used since 31P resonances in glasses are dominated by chemical shift distribution. However, a small Lorentzian contribution (ca 5%) was used to optimise the fits. 31P NMR chemical shifts are referenced to an 85% H3PO4 solution.
The 119Sn NMR spectra were recorded at 9.4 T on a Bruker AV-400 spectrometer (Larmor frequency 149.1 MHz), with a 2-μs pulse (45°) and a 60-s delay between each accumulation. A 4-mm MAS probe was used, but the spectra were recorded in static condition, since the large CSA of tin, due to its large electronic shielding, cannot be removed by MAS. 119Sn NMR chemical shifts are referenced to liquid Sn(CH3)4 (TMT).
3 Results and discussion
Table 1 reports the batch and analysed compositions of x SnO–(100 – x) P2O5 glasses. The analysed compositions are very close to the batch ones, meaning that only few losses occurred during melting (excepted for the composition x = 42). In the following text, x always refers to the analysed compositions and to the total Sn content (x = SnO + SnO2). Table 1 also reports Sn2+/Sntotal ratios. Owing to the melting under neutral argon atmosphere, the amount of Sn4+ is always low. It decreases from 10% for low x values to 1% for the highest Sn content. The solubility of Sn4+ is known to be very low in glasses [10], but no opacity was detected in the glasses for these Sn4+ contents.
Chemical and redox compositions of x SnO–(100 – x) P2O5 glasses
| SnO batch (%mol) | P2O5 batch (%mol) | SnOtot = SnO+SnO2 (%mol) ± 2 | P2O5 analysed (%mol) ± 2 | Redox Sn2+/Sntot |
| 75 | 25 | 74 | 26 | 0.99 |
| 72.5 | 27.5 | 73 | 27 | 0.98 |
| 70 | 30 | 71 | 29 | 0.99 |
| 68 | 32 | 69 | 31 | 0.97 |
| 65 | 35 | 64 | 36 | 0.98 |
| 60 | 40 | 60 | 40 | 0.95 |
| 55 | 45 | 57 | 43 | 0.95 |
| 50 | 50 | 49 | 51 | 0.93 |
| 45 | 55 | 46 | 54 | 0.94 |
| 30 | 70 | 42 | 58 | 0.9 |
Fig. 1 shows the glass-transition temperatures of x SnO–(100 – x) P2O5 glasses. Increasing x does not influence Tg before 60 mol% SnO, and only a small increase in Tg is observed above 60 mol% SnO. This is in accordance with the previously reported effect of SnO on phosphate glass properties [5]. Indeed, tin phosphate glasses are known to exhibit very low Tg values and the reticulating effect on the depolymerised phosphate network is found to be low. This effect is more evident when compared with x ZnO–(100 – x) P2O5 and x PbO–(100 – x) P2O5 glasses (Fig. 1): for both glass systems, Tg values are much larger (up to 200 K for x ZnO–(100 – x) P2O5. Surprisingly, no effect of Sn4+ is visible on Tg, whereas even small amounts of tetravalent ion like Ti4+ have a large influence of Tg [11].
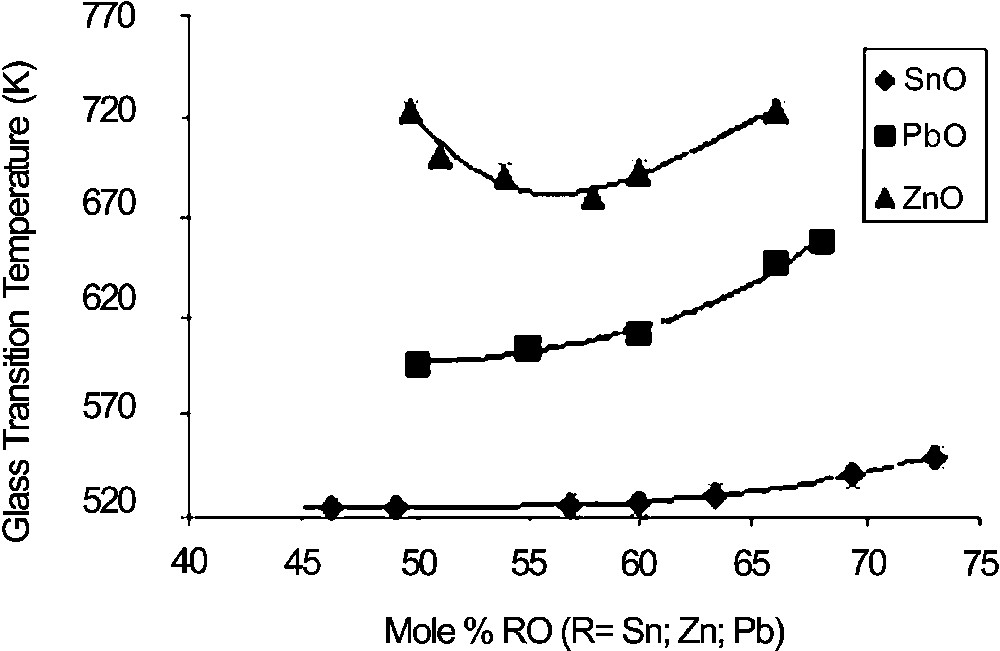
Glass-transition temperatures of RO-phosphate glasses (incertitude is within the size of symbols).
The 31P MAS-NMR spectra of x SnO–(100 – x) P2O5 glasses are shown in Fig. 2. Broad resonances are observed, in accordance with the chemical shift distribution expected for glasses. However, several resonances, attributed to different Qn sites (n = 3, 2, 1, 0), can be distinguished. The Qn assignment, shown in Fig. 2, is obtained by comparison with spectra of similar glass systems: x ZnO–(100 – x) P2O5 [12], x CaO–(100 – x) P2O5 [13], and x PbO–(100 – x) P2O5 [14]. Q3 sites are located at –43 ppm. Q2 and Q1 sites resonate between –30 and –38 ppm and between –20 and –22 ppm, respectively. The observed shifts with glass composition are attributed to a modification of the bond angle and distance distributions with SnO content in glasses [15]. Indeed, the average chain length decreases when the amount of SnO increases, which influences the bond characteristics. We notice that no specific line can be attributed to Qn sites bonded to Sn4+. Due to the higher electrostatic field strength of Sn4+ compared to Sn2+ (z/a2 = 96 and 38 nm–2, respectively), Qn sites bonded to Sn4+ are expected to resonate at higher field than those bonded to Sn2+ [11]. This absence of specific resonance may be due either to the small amount of Sn4+ (signal not detectable), or to an averaged bonding of Qn sites to both Sn2+ and Sn4+ (meaning no specific Qn site for each tin valence). This point will be discussed below.
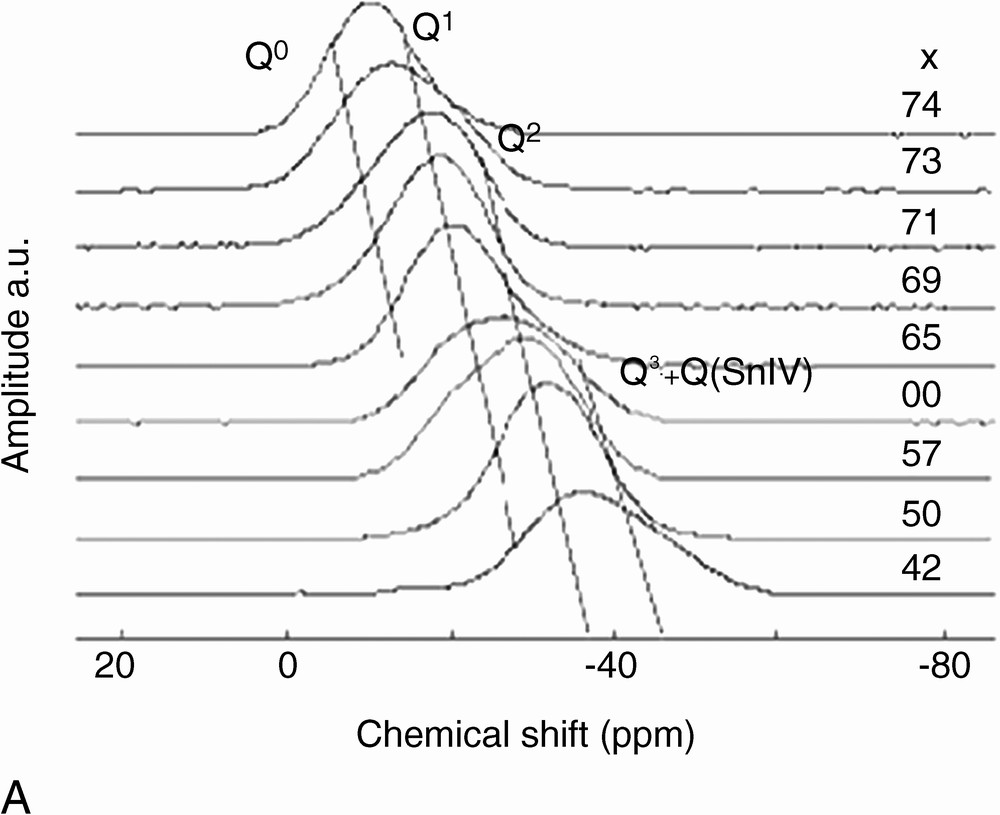
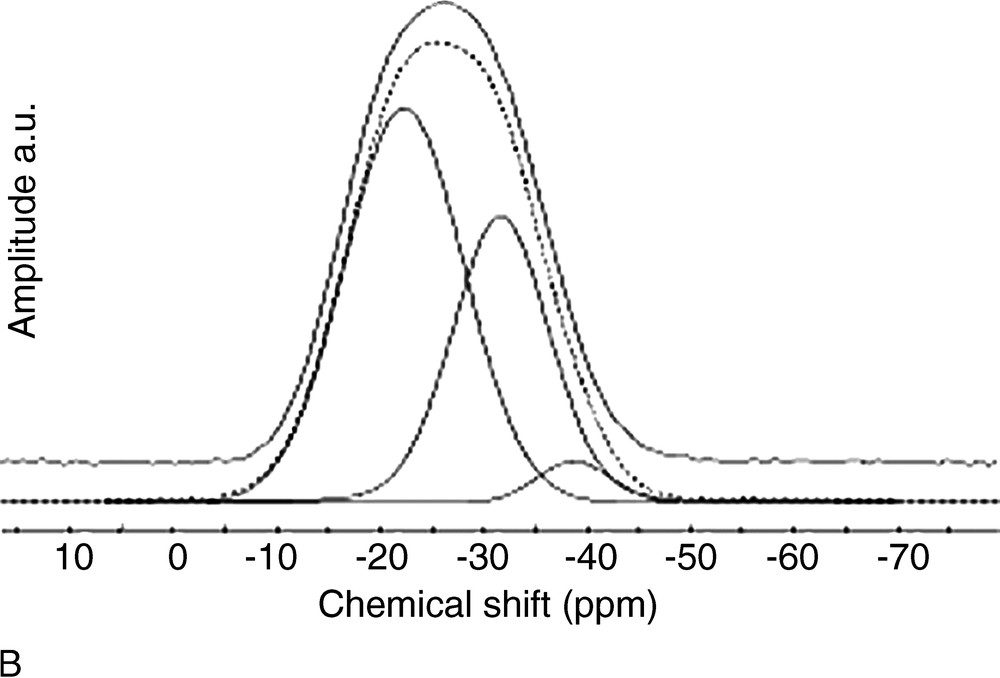
(A) 31P MAS NMR spectra of x SnO–(100 – x) P2O5. (B) Typical fit of 31P MAS NMR spectrum of 60 SnO–40 P2O5 glass. Continuous line: experimental spectrum, dotted line: calculated spectrum.
The relative amount of Qn sites can be measured by spectral decomposition with Gaussian lines, representative of chemical shift distribution encountered on glass spectra. In a first attempt for quantifying Qn sites, we neglected the contribution of Sn4+, since its effect is not visible on 31P NMR spectra. The fraction of Qn sites was calculated with the assumption of a binary Qn distribution, as generally reported in binary phosphate glasses [11–13].
If only Sn2+ is taken into account, the fractions f of the Qn species for ultraphosphate glasses (x < 0.50) is:
The fraction f of the Qn sites for polyphosphate glasses 0.50 ≤ x ≤ 0.67:
For glasses with 0.67 ≤ x ≤ 0.75:
If R = Sn2+/Sntotal is taken into account, the total QnSn species for ultraphosphate glasses (x < 0.50) is:
The fraction (f) of the Q3 sites for ultraphosphate glasses:
The calculation is the same for polyphosphate compositions.
Fig. 3 shows that there is a large discrepancy between calculated and experimental values, meaning that Sn4+ actually has a large influence on the proportion of Qn distribution. In a second step, the amount of Qn sites bonded to Sn4+ was included in the Qn site proportion, and the experimental Qn proportions were thus corrected. A further contribution was indeed included in the fits (Fig. 2b), at a chemical shift close to those of Q3 sites (ca. –38 ppm). This small resonance was neglected in the first attempt for quantifying Qn sites, but its presence enables to obtain a better fit, and its small intensity is actually in accordance with the small amount of Sn4+ in the glasses (Table 1). Fig. 3 shows that the corrected Qn proportions are in very good accordance with the calculated values.
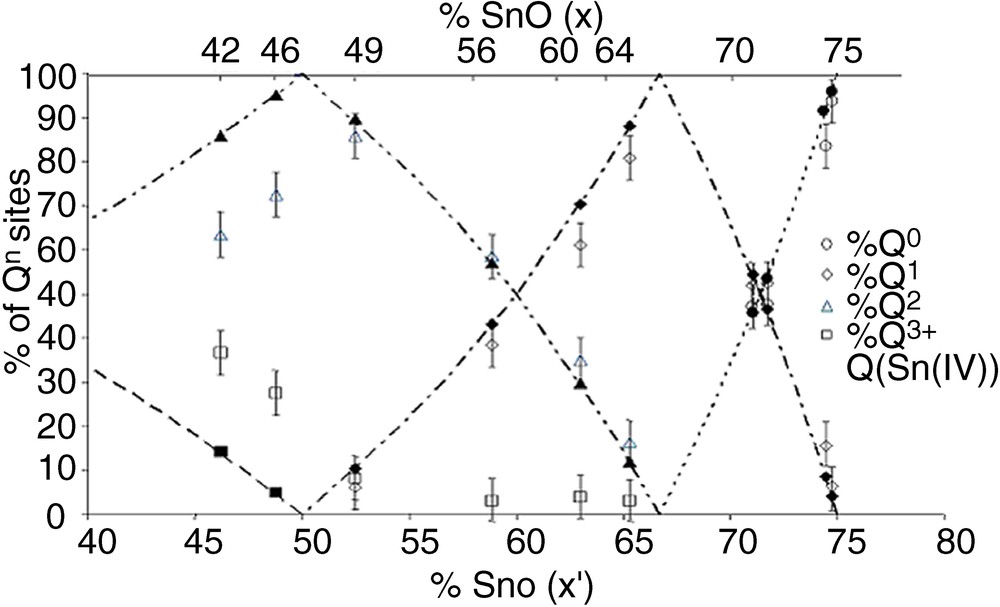
Qn fractions of x SnO–(100 – x) P2O5 glasses versus x (top scale), or versus x′ = SnO + 2 × SnO2 (bottom scale). Dotted lines: calculated values. Open symbols: experimental values obtained from fitting of NMR spectra, without contribution of Sn4+. Closed symbols: experimental values obtained from fitting NMR spectra with contribution of Qn sites bonded to Sn4+(Q(Sn(IV)).
We notice that further information on Qn sites connectivity may have been obtained with double-quantum filtered 31P MAS–NMR [12,16], or even better with the recently reported INADEQUATE filter which probes scalar couplings [17]. However, the resolution of Qn sites on our 31P MAS–NMR spectra is not enough to obtain separate cross peaks.
Tin NMR spectra are dominated by a very large CSA (Fig. 4), which cannot be removed by magic angle spinning (spectra width is larger than 95 kHz). This behaviour is similar to that observed for 207Pb NMR, although static tin NMR spectra keep a well-defined line shape contrary to 207Pb spectra. The simulation of static spectra enabled to measure the chemical shift anisotropy span (Ω) and to estimate the averaged value of isotropic chemical shift (which takes into account the chemical shift distribution). Unfortunately, there are very few chemical shift data for inorganic tin compounds and even less for tin phosphates. Only three crystalline tin phosphate compounds are known: SnP2O7 (Q1, Sn4+), Sn2P2O7 (Q1, Sn2+) and Sn3(PO4)2 (Q0, Sn2+). Curiously, no crystalline tin metaphosphate is known [18]. Moreover, the crystal structure of Sn2P2O7 is not reported. Fig. 5 shows NMR data for these compounds. Some trends can be proposed: by similarity with lead NMR [17] and by examining data in Fig. 5, increasing the covalent character of Sn–O bonds will induce an increase in the CSA span, and a down-field shift of the static line. Fig. 5 illustrates the evolution of CSA span and δiso for 119Sn spectra of x SnO–(100 – x) P2O5 glasses. The isotropic chemical shifts (δiso) reported in Fig. 5 were obtained by recording 119Sn MAS–NMR spectra at a very low field (2.34 T) and a spinning rate of 15 kHz, which enabled to observe isotropic lines resolved from spinning side-bands. They were also estimated by fitting the static spectra recorded at 9.4 T.
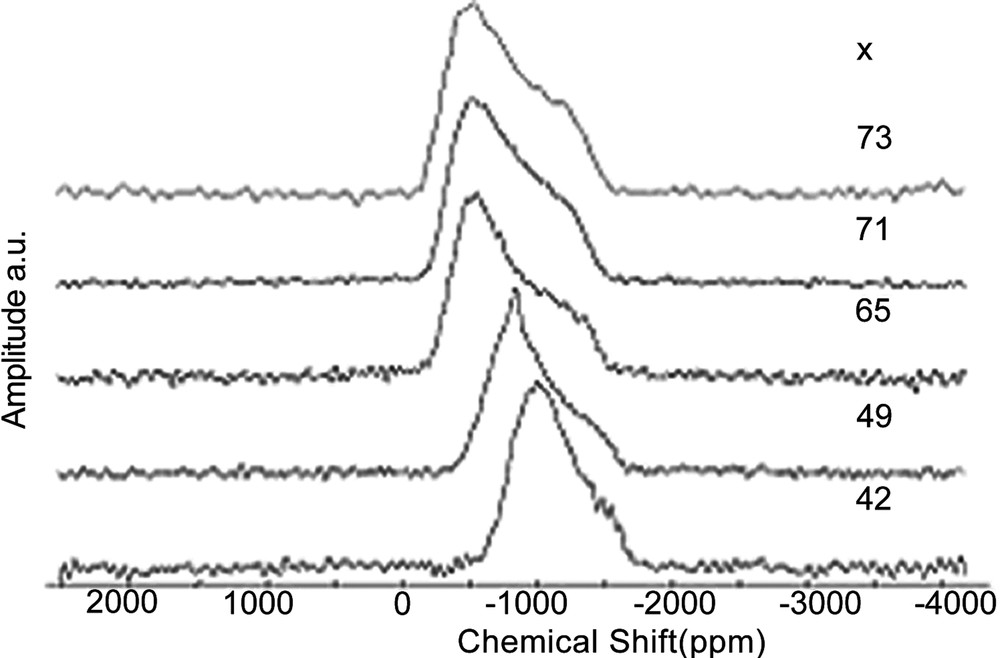
Static 119Sn spectra of x SnO – (100 – x) P2O5 glasses.
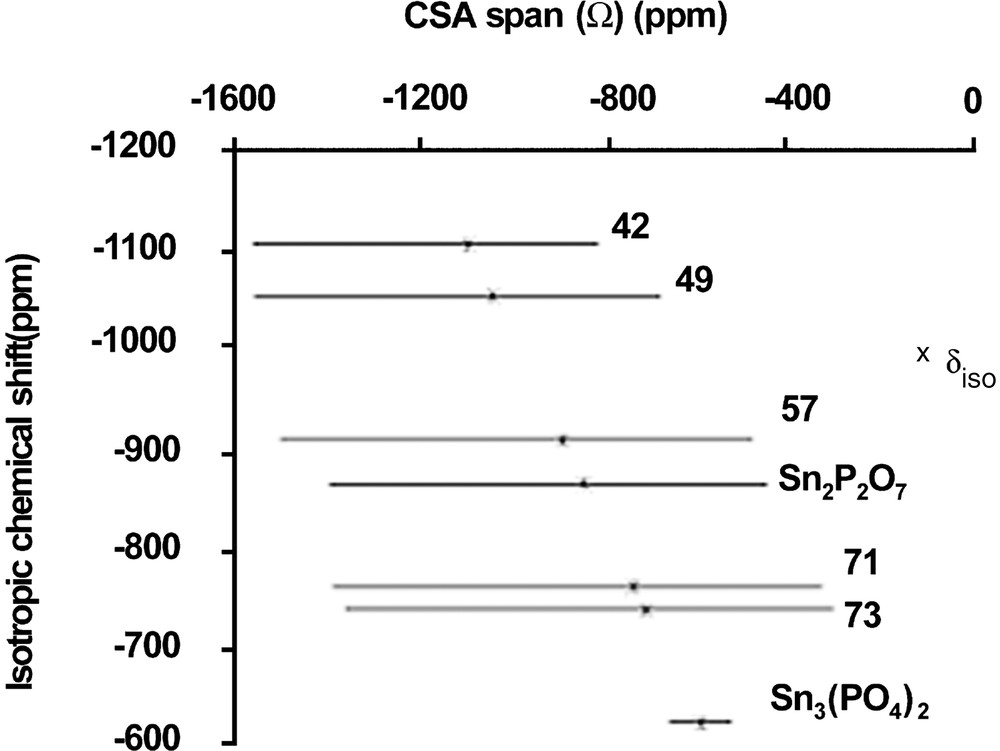
Plot of 119Sn NMR data for some crystalline reference compounds and x SnO–(100 – x)P2O5 glasses.
When the SnO amount increases in the glass, tin is bonded to less and less polymerised Qn sites (Q2 down to Q0), meaning that Sn–O bonds become more covalent. This is in accordance with the evolution of CSA span and δiso observed in Fig. 5. Moreover, Fig. 5 shows that the spectra of Sn2P2O7 and Sn3(PO4)2 are close to those obtained in glasses, excepted for the lower CSA span of Sn3(PO4)2, due to the absence of distribution. We conclude that in glasses containing a high amount of tin(II), the tin(II) environment should be close to that observed in Sn3(PO4)2, i.e. a trigonal pyramid. The presence of Sn4+ is detected only on the static tin NMR spectrum of x = 49 glass, probably because of its small amount in the other glasses. Like in silicate and germanate glasses, a narrow resonance near –800 ppm indicates the presence of Sn4+ with a symmetric environment, generally octahedral in crystalline tin(IV) compounds. We notice that the chemical shift and CSA are close to those obtained in crystalline SnP2O7, suggesting that Sn4+ in the glasses has an octahedral environment similar to that in crystalline SnP2O7.
4 Conclusion
Although even small quantities of tetravalent ions have large effects on glass structure and properties, the presence of Sn4+ in x SnO–(100 – x) P2O5 is difficult to detect on 31P and 119Sn spectra, as well as on glass transition temperatures. The influence of Sn4+ becomes visible only for the most oxidised glasses in which the Sn4+ local environment is close to SnP2O7. This suggests that Sn4+ may segregate as SnP2O7 even in non-oxidised tin phosphate glasses.
Acknowledgements
E.B. is grateful to the ADEME for providing a financial support to its thesis work, and A. Kunegel (ADEME, Angers) for its technical support.