

1 Introduction
Wells have been drilled for different purposes: exploration, disposal, oil and gas production… All these wells will be abandoned and will require plugging. The goal of each well abandonment should be to control any leakage of fluids within the well to avoid the risk of contamination of freshwater aquifer and to monitor the state of well sealing in order to prevent surface or sea pollution [1]. Many formulations have been developed for oilwell cementation; however, only few studies have been devoted to the physical and chemical processes causing cement degradation under downhole conditions [2–5].
The objective of this work is to study the structure and the properties of cement formulations cured and stored under different conditions of temperature and pressure. The XRD allows us to investigate the highly crystallized phase, whereas NMR is very adapted to the study of such heterogeneous and multiple phase material [6]. It is also one of the only available techniques able to investigate the calcium silicate hydrates (C–S–H), which are the main constituents of the Portland cement paste. It is well known that C–S–H present a poorly crystalline structure and variable composition [7].
2 Experimental details
Two cement formulations, a class G Portland cement (labelled C in the text) and a Low Permeability Cement (LPC), with two curing conditions were prepared for this study as described in Table 1. The unhydrated cement used was a class-G Portland cement from the Dyckerhoff Company (Bogue composition (wt%): 51.2 Ca3SiO5, 27 Ca2SiO4, 2.3 Ca3Al2O6, 14.4 Ca4Al2Fe2O10). In the case of LPC samples, sand and silica fume were added to the class-G Portland cement to increase mechanical and durability properties by optimising the material compacity [8]. The addition of silica fume produces secondary hydrates by pozzolanic reaction with the lime resulting from primary hydration (SiO2 + Ca(OH)2 → C–S–H) [9]. Silica fume has a very high water demand, because of its high specific-surface area, so a cement-dispersant, polynaphtalene sulfonate (PNS), was incorporated in the mixes to maintain adequate consistency at a reasonable water/cement ratio.
Characteristics of cement mixes (weight ratios relative to cement mass) and curing conditions
| Formulations | LPC | C | ||
| Components | LPC I | LPC II | C I | C II |
| Cement class G | 1 | 1 | ||
| Silica fume | 0.24 | — | ||
| Sand | 0.2 | — | ||
| Cement dispersant | 0.018 | — | ||
| Water | 0.27 | 0.44 | ||
| Curing conditions (30 days in tap water) | T = 293 K | T = 353 K | T = 293 K | T = 353 K |
| p = 105 Pa | p = 7 × 106 Pa | p = 105 Pa | p = 7 × 106 Pa |
X-ray diffraction (XRD) data were collected using a Philips PW 1820 diffractomer employing the Co–Kα radiation (λ0 = 1.789 Å). The samples were scanned at 0.6° per minute between 2 and 82° 2θ.
The 27Al and 29Si NMR experimental details are reported in Table 2. Single-pulse experiments were carried out in order to respect the relaxation times of the species present in the samples except for sand. The 27Al and 29Si chemical shifts were respectively referenced relative to a 1.0 M AlCl3–6 H2O solution and to tetramethylsilane Si(CH3)4 (TMS) at 0 ppm, using Si[(CH3)3]8Si8O20 (Q8M8) as a secondary reference (the major peak being at 11.6 ppm relatively to TMS).
Experimental details for NMR measurements
| Nucleus | Sample | Spectrometer Bruker | ZrO2 rotor | Frequency (MHz) | Pulse width | Relaxation delay (s) | Spinning rate (kHz) |
| 27Al | LPC, C and raw materials | ASX 500 | 2.5 mm | 129.80 | π/12 (0.5 μs) | 1 | 25 |
| 29Si | LPC | 11.7 T | 7 mm | 99.305 | π/2 | 60 | 5.5 |
| C and raw materials | ASX 300 | 4 mm | 59.591 | 3 | 7 | ||
| 7.05 T |
3 Results and discussion
3.1 X-ray diffraction
Phases identification of hardened cements after a 30 days-cure at ambient temperature and atmospheric pressure by XRD analysis (Fig. 1) shows the presence of unhydrated phases alite Ca3SiO5, belite Ca2SiO4, ferrite phase Ca2(AlxFe1–x)2O5 (where 0 < x < 0.7) along with α-quartz due to siliceous aggregate used in LPC samples and usual hydrated phases such as portlandite Ca(OH)2, and ettringite [Ca3Al(OH)612 H2O]2(SO4)32 H2O (AFt phase). In the samples cured at high temperature and high pressure, ettringite phase was absent. In the CII sample, instead ettringite, hydrogrossular CaO3Al2O3(SiO2)3–x (H2O)2x (where x = 0 to 3) have been formed. Under conditions of elevated temperature, previous studies have shown that hydrogrossular Si-free will form, which is the most thermodynamically stable and the least soluble of the calcium aluminate hydrates [10]. In the LPCII spectra, all peaks characterising the portlandite phase [Ca(OH)2] have completely disappeared. The calcium hydroxide released during cement hydration is actually consumed as a result of interaction with active silica fume to form C–S–H phases.
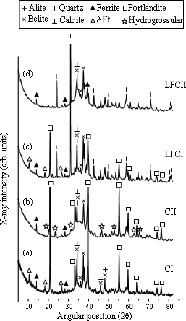
XRD patterns of samples hydrated for 30 days at (a, c): T = 293K, p = 105 Pa and (b, d): T = 353 K, p = 7 × 106 Pa, Co Kα radiation.
3.2 Magic Angle Spinning Nuclear Magnetic Resonance Spectroscopy
3.2.1 29Si MAS NMR
An example of the spectra decomposition is presented in Fig. 2d, where we have also reported NMR spectra of the raw materials (Fig. 2a–c). It is important to note that even though the presence of Fe can cause peak broadening and the Fe-content of oilwell Portland cement is quite large, reasonably well-defined spectra can be obtained. The 29Si NMR spectrum in Fig. 2a displays a narrow line of Q4 type at –107 ppm and is characteristic of crystalline quartz, whereas the broad peak at –110 ppm in the spectrum for silica fume (Fig. 2b) is characteristic of amorphous SiO2. Fig. 2c shows the 29Si NMR spectrum of class-G cement. It contains a broad Q0 component near –71 ppm, which is the sum of alite and belite. The observed line broadening arises from the incorporation of metal (Mg2+, Al3+ and Fe3+) and other impurity ions into the crystal lattice [11]. All raw materials are still present in the hydrated cement (Fig. 2d) with usual non-stoichiometric and non-crystalline calcium silicate hydrates (C–S–H). The main resonance lines at –79.2 ppm and –85.5 ppm are respectively due to the end-chain tetrahedra Q1 and nonbridging tetrahedra Q2 of the C–S–H [12]. The peak for Q2 sites is asymmetric or has a small shoulder at about –82.5 ppm. In calcium silicate hydrate, this peak has been already assigned to the middle tetrahedral of the dreierkette C–S–H chain structure (Q2L) [13, 14], to distorted Q2 sites produced by various kinds of staking disorder [15] or to Q2 species originating from precipitation of hydrate from the small quantities of bleed water resulting from the large centrifugal forces induced by the MAS technique [16]. Furthermore, in our samples, the C–S–H can also contain Al and the Q2(1Al) are expected in the same shift range as the Q2L [17]. Thus, assignment of this peak is a difficult problem that remains unresolved. The Q3 peak observed only as a shoulder near –90 ppm is ascribed to the cross-linked C–S–H structure [18].
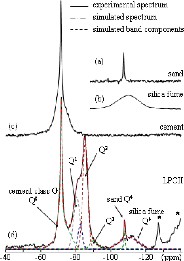
29Si MAS NMR spectra of raw materials: (a) sand, (b) silica fume, (c) cement, and (d) LPCII sample hydrated for 30 days at T = 353 K and p = 7 × 106 Pa with its deconvoluted peaks. The asterisks mark spinning sidebands.
The 29Si MAS NMR spectra of C and LPC samples are presented in Fig. 3. As temperature and pressure increase, the relative intensity of the Q0 peak decreases. This is linked to an acceleration of the hydration kinetics with temperature and pressure, the Q0 anhydrous species being transformed into calcium silicate hydrate (C–S–H). Furthermore, the ratio Q2/Q1 increases with temperature and pressure. In addition, there may be some Q3 sites present in the treated samples. These results indicate that increasing temperature and pressure increases the polymerisation of the C–S–H structure. Such structural changes have been previously observed on hydration of tricalcium silicate at high temperatures and high pressure [19].
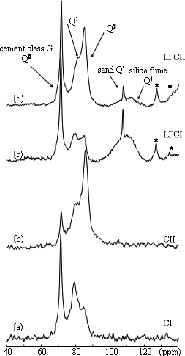
29Si MAS NMR spectra of samples hydrated for 30 days at (a, c): T = 293 K, p = 105Pa and (b, d): T = 353 K, p = 7 × 106 Pa. The asterisks mark spinning sidebands.
In the case of LPC samples, we can observe an important decrease of the relative intensity of the silica fume peak with increasing temperature and pressure. This phenomenon can be attributed to the progression of the pozzolanic activity of silica fume and is in good accordance with the disappearance of portlandite in XRD spectra of the LCPII sample. This result is in agreement with studies by Zanni et al. that indicate a weak and low activity of the silica fume at ambient temperature, which increases with temperature [20].
3.2.2 27Al MAS NMR
The main part of the bulk Al2O3 content in Ordinary Portland Cement is present in the so-called interstitial material, which contains the aluminate and ferrite phases. However, for oilwell cements that contain a low bulk Al2O3:Fe2O3 ratio, the aluminate phase is usually absent or present in very small quantities and Al is mainly in the ferrite phase [21].
The 27Al NMR spectrum of anhydrous cement (Fig. 4a) shows a broad asymmetric band of intensity near 80 ppm corresponding to tetrahedrally coordinated Al. The peaks near 13 ppm and 10 ppm are attributed to Al in octahedral sites. The coordination environment of aluminium in anhydrous cement is variable; anhydrous cement may contain both octahedral and tetrahedral sites or only tetrahedral sites [11,18]. However, octahedral sites observed may be assigned to hydration product during storage. Skibsted et al. [22] provides compelling evidence that the resonances in the tetrahedral region arise from Al for Si substitution in the alite and belite phases. They also show that Al present in the tetracalcium aluminoferrite phase of cement, either in the antiferromagnetic or the paramagnetic form, contributes little or no to the observed 27Al MAS NMR spectrum [23]. On reaction with water, it initially forms ettringite [Ca3Al(OH)612 H2O]2(SO4)32 H2O (AFt phase) and later the thermodynamically stable monosulfoaluminate [Ca2Al(OH)6]2(SO4)12 H2O (AFm phase), which all exclusively contain octahedrally coordinated Al [24] and leads to peaks in 27Al NMR spectra respectively at 13 and 10 ppm [17,22,25]. We can notice that AFm phases are not detected by XRD, indicating that the AFm material is poorly crystalline. Taylor has suggested that AFm phases could be incorporated within the C–S–H, perhaps within or near the silicate layers [24]. The peak attributed to AFm is present in all our samples but the AFt peak is not or weakly present in treated samples CII and LPCII.
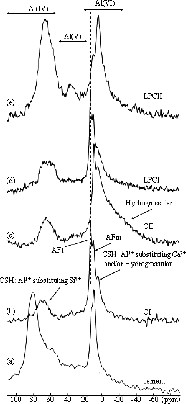
27Al MAS NMR spectra of (a) anhydrous cement and samples hydrated for 30 days at (b, d): T = 293 K, p = 105 Pa and (c, e): T = 353 K, p = 7 × 106 Pa.
We can also notice the presence of a band at 4 ppm, particularly intense in the LPCII sample and a broad band near –20 ppm in the CII sample. The broadest line near –20 ppm has been attributed to hydrogrossular phases [17]. The important width of this band can be explained by the highly distortion of site because of Si-site partial substitution by OH and Al-site by iron. This attribution is in agreement with XRD results that show the presence of hydrogrossular phases in the CII sample. The attribution of the peak at 4 ppm is not clear and has been also assigned to hydrogrossular [25], but in the investigations of aluminium incorporation in the C–S–H, this peak has been assigned to Al3+ substituting Ca2+ in the octahedral sheet of the C–S–H structure [26]. However, as Taylor pointed out, the large difference in ionic radius between Al3+ and Ca2+ makes unlikely that Al3+ would replace Ca2+ randomly. Taylor proposed that Al replacement of Ca2+ could only occur with the formation of AFm or other phases in which Al is octahedrally coordinated, leaving the C–S–H with very limited substitution [24].
In sample LPCII, a band is clearly detected near 36ppm and tentatively assigned by Faucon et al. to pentacoordinated Al3+ substituting for Ca2+ ions situated in the interlayers of the C–S–H structure [26]. The MAS spectra of the hydrated samples (Fig. 4b–e) also yield signal in the Al(IV) range particularly intense in the LPCII sample, but the peak maximum has changed to about +65 ppm from +82 ppm for the unhydrated cement. According to Skibsted et al. [23], the more shielded peak may arise from Al incorporated in the C–S–H.
4 Conclusion
Two cement formulations with different curing conditions were analysed by XRD and NMR measurements. The raw materials are still present in all the samples hydrated one month. The increase of pressure and temperature lead to a more polymerised C–S–H. In the cement C, we observed an acceleration of the hydration kinetics with temperature and pressure. The hydrated phases of the CI sample consisted of poorly crystalline C–S–H, AFm with AFt and portlandite. In the sample cured at 353 K, 7 × 105 Pa, AFt phase is absent; hydrogrossular had formed instead. In the LPC samples, it was shown that the pozzolanic activity of silica fumes increases with temperature and pressure and lead to the consummation of all the portlandite released during cement hydration. This feature is quite important and positive for long-term durability aspects. Indeed, it is well known that calcium hydroxide is easily soluble as soon as pH is lower than 12.5 at room temperature. In order to have a better characterisation of the minor phases present in the cement, selective dissolution is underway. Furthermore, mechanical tests and porosity measurements are in progress to try to establish a correlation between the structure and macroscopic properties.
Acknowledgements
The authors would like to thank ‘Institut français du pétrole’ for the permission to publish this paper. They are grateful to Annie Audibert for valuable discussions, and Bernadette Rebours, Sylvie Massot, Isabelle Clémençon for their help in XRD measurements.