

In 1995, Yamada and co-workers isolated Dolabelides A and B, two 22-membered ring lactones, from the sea hare Dolabella auricularia (family Aplysiidae) [1]. In 1997, two similar 24-membered ring lactones, Dolabelides C and D, were also extracted from the same source [2]. These compounds were shown to exhibit cytotoxicity against HeLaSe3 cell lines with IC50 values of 6.3, 1.3, 1.9, and 1.5 μg ml–1, respectively. Their structures were determined by HRFAB mass spectroscopy and 2D NMR, and their absolute configuration by the modified Mosher method [3]. Several groups have reported syntheses of Dolabelide fragments (C16–C24: [4], C15–C24 and C25–C30: [5], C15–C30: [6]).
The retrosynthesis that we envisioned is illustrated in Fig. 1. Opening the macrolactone and disconnecting the C15-C16 bond furnishes two fragments of roughly equal sizes, C1–C15 and C16–C30. They would be coupled by a B-alkyl Suzuki reaction between the vinyl iodide at C15 and a borane derived from the olefin at C16 (for a recent use of such a coupling reaction, see [7]). The C15–C30 portion can be further disconnected through the C24–C25 double bond. In a previous paper, we described the synthesis of C16–C24 aldehyde 1 [4], which could be engaged in a Wittig coupling with phosphorane 2. An alternative to make the C24–C25 bond would be a Julia coupling between β-hydroxy sulfone 3 and ketone 4.
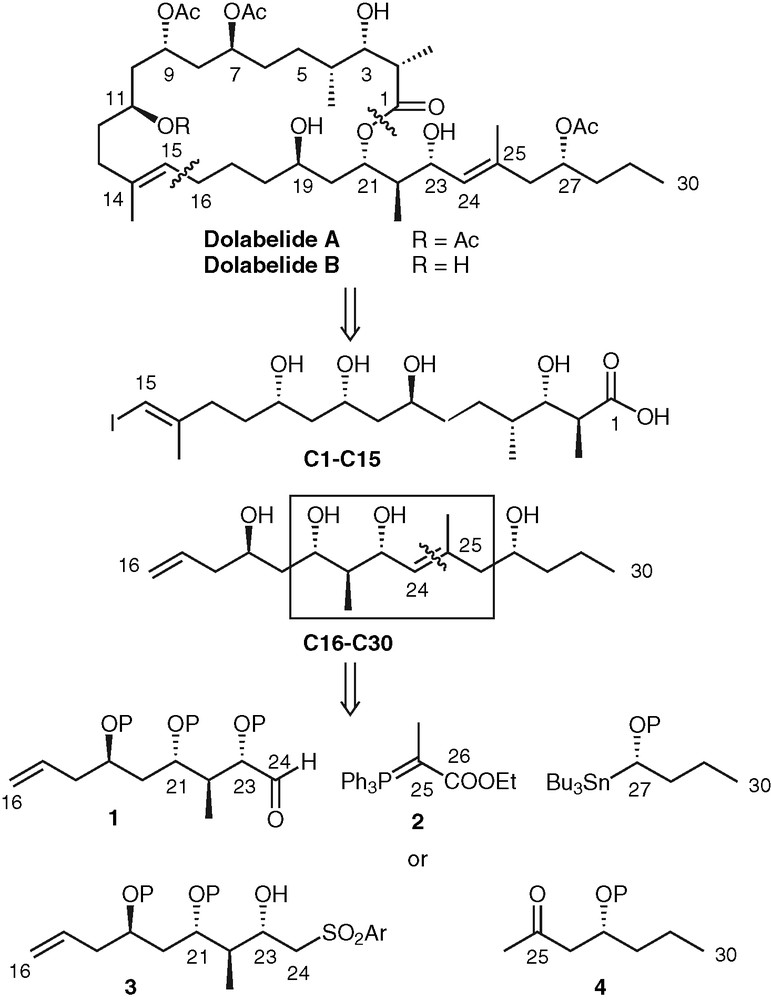
Dolabelide A retrosynthesis.
Model hydroxy sulfones 7a–b were easily prepared in two steps from vinyl sulfones 5 according to previous work in our laboratory [8]. Intramolecular conjugate addition of an intermediate hemiacetal anion made in situ from homoallylic alcohols 5a-b with benzaldehyde and potassium tert-butoxide gave the protected syn 1,3-diols 6a-b. Regioselective reduction of these benzylidene acetals with DIBAL-H furnished the corresponding hydroxy sulfones 7a–b in good yields (Fig. 2).
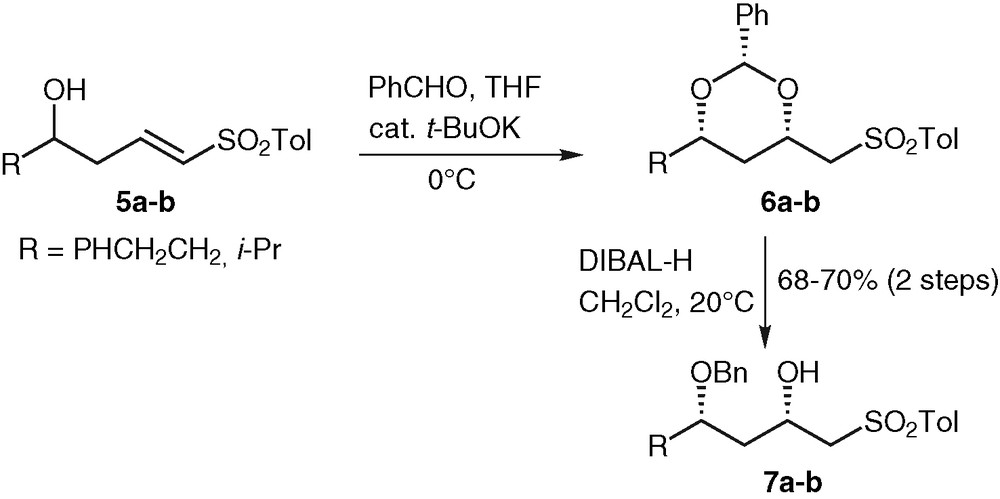
Synthesis of model β-hydroxy sulfones.
First attempts of condensation of the dianions of sulfones 7a and 7b according to a literature procedure [9,10] gave modest yields of the desired diols 8 (from 23 to 37% when using isobutyraldehyde, and 59% for benzaldehyde) due to the poor conversion of the starting sulfones (Fig. 3) [8]. Several additives were employed to try to improve the conversion of sulfones 7a–b: LiBr, HMPA, TMSCl or AcCl, with no success. Other bases were screened: BuLi/t-BuOK, LDA, Et2NLi gave similar results, and the conversion did not exceed 12% with i-PrMgCl. A possible explanation for the poor conversion could be the enolisation of the carbonyl compound, although no aldol side-products were observed.

First attempts of condensation of β-hydroxy sulfones with aldehydes.
Finally, PhLi·LiBr proved to be the base of choice. This reagent was first utilized by Masamune for a Julia coupling during the final steps of the synthesis of bryostatin 7 [11]. Yields improved to 67–80%, and the yields based on recovered sulfones were excellent (up to 95%). Moreover, only 1.2 equivalent of aldehydes can be used for optimum results. The crude adducts were directly transformed into the corresponding acetonides for two purposes: easier separation of the products from the starting hydroxy sulfones, and determination of the relative stereochemistry of the newly formed centres. Four diastereomers were observed in all cases, and their configuration was proved by 1H [9] and 13C NMR analysis [12–14]. Tanikaga et al. reported the formation of only two diastereomers for the condensation of simpler β-hydroxy sulfones with similar aldehydes [10]. They correspond to the major isomers in our case (compounds 9 and 10). We have no explanation for the discrepancy between the selectivities in our study and in Tanikaga's report (Fig. 4).
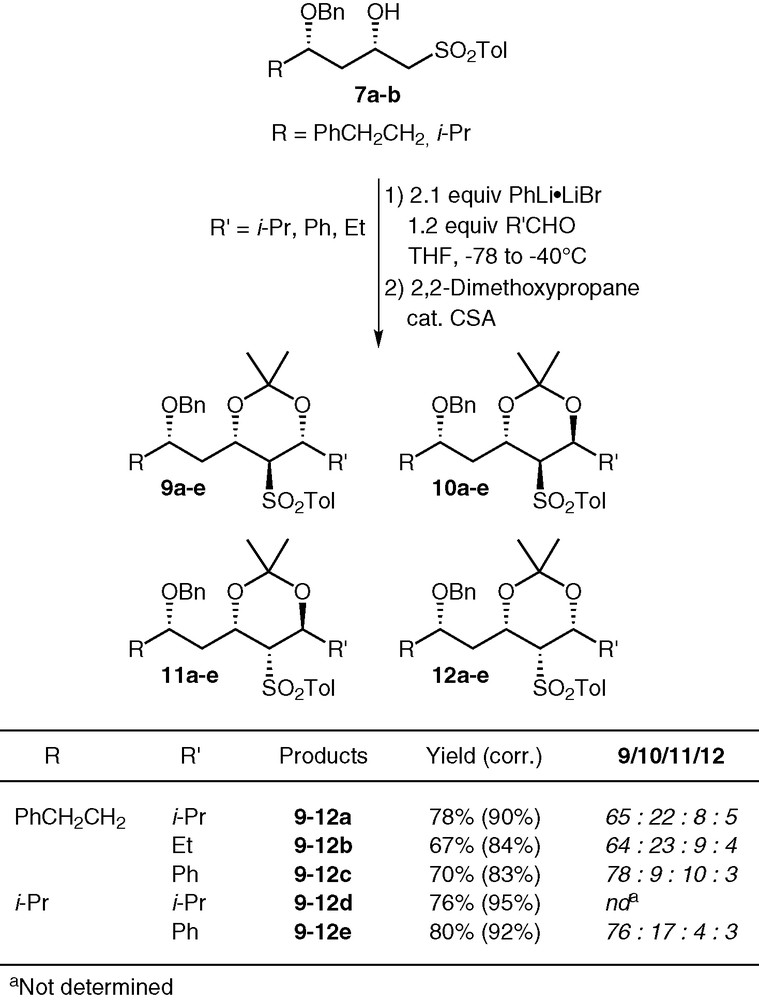
Condensation with PhLi as base.
Addition of dianions of β-hydroxy sulfones to ketones has also been reported [15]; so we tried the condensation of sulfone 7a with acetone (Fig. 5). The yield of this reaction is not as satisfying as with aldehydes, but the selectivity is comparable.
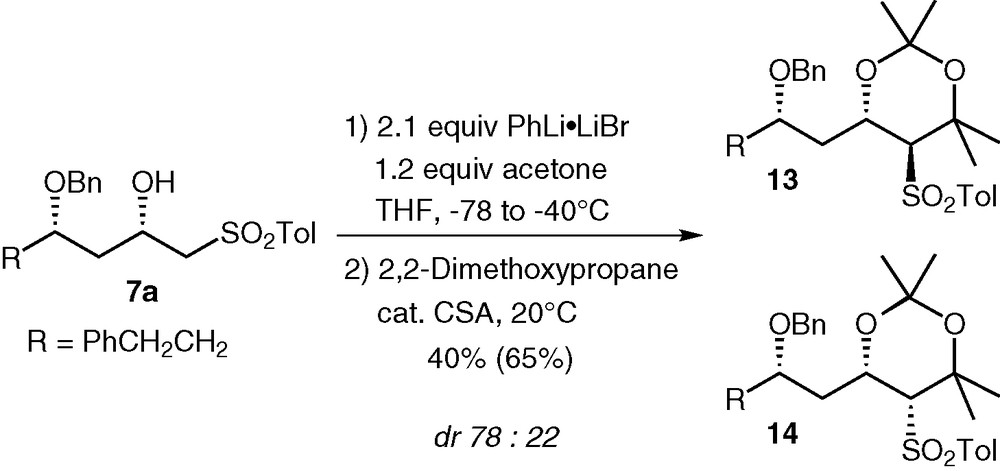
Condensation of sulfone 7a with acetone.
Since we had difficulties coupling sulfones 7 with aldehydes or ketones at the beginning of this study, we examined an alternate route to model compounds of the C16–C30 portion of Dolabelides, featuring a syn 1,3-diol unit flanked by an olefin (see the boxed portion of C16–C30 in Fig. 1). We envisaged creating the C24–C25 before performing the conjugate addition that installs the syn diol functionality. Hydroxy vinyl sulfones 5 (R = PhCH2CH2, Ph, C4H9) were protected as the corresponding tert-butyldimethylsilyl or triethylsilyl ethers, leading to compounds 15a–c in quantitative yield (Fig. 6). Deprotonation of sulfone 15a (R = PhCH2CH2) was first attempted with tert-BuLi, followed by addition to isobutyraldehyde. The reaction was clean, but 16a was obtained in only 40% yield. Here again, PhLi·LiBr solved this problem, and sulfones 16a-d were formed in good to excellent yields (64–83%) as 1:1 mixtures of diastereomers (Fig. 6).
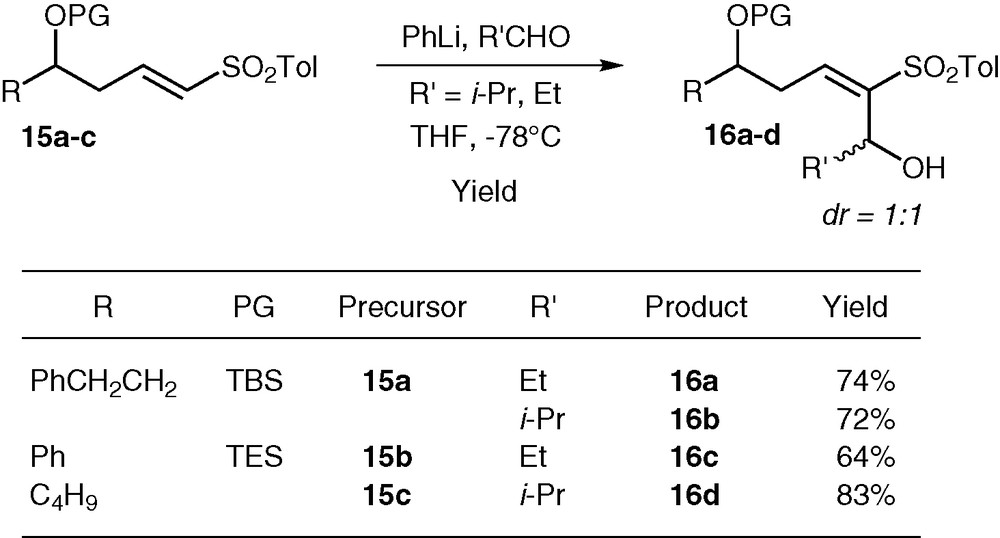
Alternate route to models of the C16–C30 portion of Dolabelides.
The reason why PhLi gives such good results with both hydroxy sulfones and vinyl sulfones is not entirely clear. It is slightly less basic than BuLi, so deprotonation of the aromatic protons ortho to the sulfone group is less favoured [16]. On the other hand, PhLi is less prone to monoelectronic transfers that might reduce the sulfonyl group.
In order to perform the conjugate addition on compounds 16, we needed to deprotect the silyl ether (PG), after having protected the alcohol group to prevent it from interfering in the 1,4-addition. At this point, we surmised it would be possible to install the syn 1,3-diol and the double bond at the same time, by activating the alcohol instead of protecting it. The anion formed from 17 after the conjugate addition would undergo elimination with the neighbouring activated group, leading to vinyl sulfone 18 (Fig. 7). In this case, a full equivalent of base would be necessary to drive the reaction to completion.
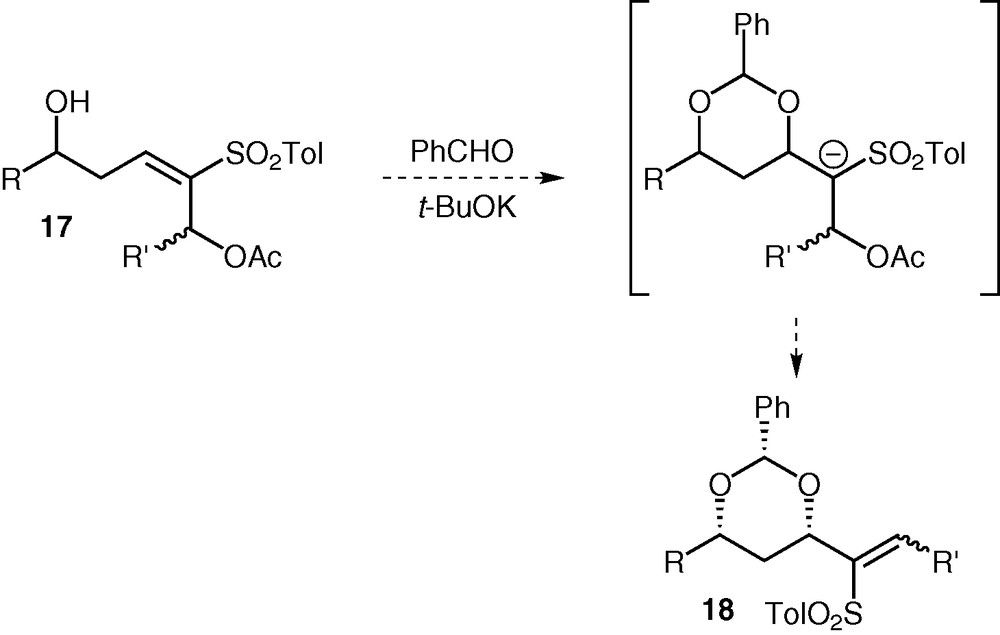
Mechanism of the conjugate addition/elimination sequence.
To verify this hypothesis, we transformed the alcohol function of 16a into an acetate (80%), and deprotected the silyl ether with a 5:95 aqueous HF/acetonitrile solution (92%). The resulting compound 17 was treated with excess benzaldehyde and a stoichiometric amount of potassium tert-butoxide. We were delighted to see that benzylidene acetal 18 was obtained in 65% yield, and with a syn/anti selectivity of 93:7 (Fig. 8). The conjugate addition is under thermodynamic control (by analogy with the conjugate addition involving unsaturated esters [17]), leading to the protected syn 1,3-diol, where all the substituents are equatorial on the benzylidene ring. The fact that we observed excellent syn/anti selectivity in the tandem conjugate addition/elimination means that the thermodynamic equilibrium is reached before the subsequent irreversible elimination takes place. We are currently studying this sequence, and especially the relation between the ratio of diastereomers of 17 and the E/Z selectivity. Efforts towards the reduction of the sulfone moiety in compounds 9-12 and 18 are also in progress.
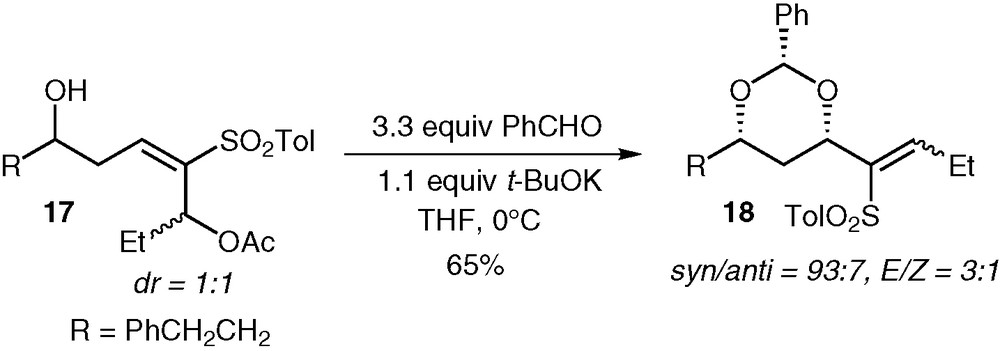
First attempt of conjugate addition/elimination sequence.
In summary, we have shown that PhLi·LiBr is an efficient base for the condensation of both β-hydroxysulfones and vinyl sulfones with aldehydes, and we designed a short route to syn 1,3,5-triols and to syn 1,3-diols bearing an olefin on the α carbon, which are model compounds for the C21–C25 sub-unit of Dolabelides.
Acknowledgments
D.R.-S. acknowledges the MENR for a fellowship. Financial support was provided by the CNRS (UMR 7652) and the ‘École polytechnique’. Pr. Jérôme Lacour is thanked for helpful discussions.