

1 Introduction
Hydrogen, as a source of energy for stationary power plants and moving vehicles, will almost certainly be widely used in a decade or so and is now in trials in several cities world-wide [1,2]. Several sources for hydrogen are now considered. Methanol, ethanol, and natural gas are the most important candidates for an efficient production of hydrogen. Ethanol has a major advantage when compared to the other two candidates: it can be easily made from renewable sources. Ethanol can be produced either by hydrolysis of cellulosic materials followed by fermentation or by direct fermentation of sugar containing corps. In both processes a mixture of ethanol-water with a ratio close to 1 ethanol for 10 water molecules is formed. It is thus highly desirable to find efficient catalytic materials for the production of hydrogen in a high water to ethanol ratios. We have previously investigated the hydrogen production from ethanol by direct oxidation (Rh–Pt/CeO2 [3] and Au/CeO2 [4]) or via reforming with water (Rh/CeO2–ZrO2 [5]). An important requirement for the catalytic hydrogen production from any of the carbon-containing source is the absence of CO. In order to power, with hydrogen, moving or stationary plants, fuel cells are used. The irreversible adsorption of CO on the Pt anode of the fuel cells at the operating temperature (about 80 °C) degrades their performance and ends by totally poising them. Thus, the working catalyst is required to be both very efficient for abstracting hydrogen atoms from the carbon containing reactant and fully oxidising the carbon atoms to CO2.
Among the good supports for oxidising CO to CO2, cerium oxide has been shown as one of the most efficient [6,7], and the addition of ZrO2 to CeO2 has resulted in enhancing the ceria activity for oxidation reactions [8,9]. This enhancement has been reported as due to increasing oxygen mobility and the formation of a solid solution.
In the case of making hydrogen from ethanol, a noble metal capable of breaking the carbon-carbon bond is needed. Rh/CeO2 [10,11] and Rh/Al2O3 [12,13] (alone or in presence of a second metal: Pd or Pt) catalysts have shown interesting activity for ethanol decomposition in presence of oxygen or water.
In this work we show that total conversion of ethanol can be achieved at reasonable temperatures with a hydrogen selectivity of 70 mol% over Rh catalysts. Moreover, catalysts composed of both Ce and Zr oxides gave the highest CO2/CO ratios.
2 Experimental
The method for making the catalysts has been reported previously and only a brief description is given here. CeO2, ZrO2, and CeO2–ZrO2 supports were prepared by precipitation with an ammonia solution of cerium and/or zirconium nitrates (Strem). Rh (2 wt%) was added by impregnation of an aqueous solution containing RhCl3
Catalytic reactions of ethanol reforming were conducted in a fixed-bed reactor (containing 100 mg of catalyst) with a molar ratio H2O/ethanol = 8, a flow rate of 0.77 l h–1 and an Ar flow rate of 3 l h–1 (Ar/H2O/ethanol = 35/8/1). Prior to the catalytic reaction, all catalysts were reduced in a H2 flow (10% in Ar) at 320 °C (1 h). Reaction products were analysed as a function of time at different temperatures. The results reported here are those taken after 90-min reaction (a steady-state condition was usually obtained after ca. 30 min). Products were analysed using three GC apparatus, equipped with FID and TCD.
X-ray photoelectron spectroscopy (XPS) was conducted in a UHV chamber, using a double pass CMA Phi 20-805 analyser and a Phi 32-095 X-ray source at the following conditions: Mg Kα for X-rays at 300 W, 50 eV pass energy for all elements, 0.1 eV/step and 300 ms/step.
3 Results and discussion
BET surface area of ZrO2, CeO2–ZrO2 (Ce/Zr = 4, 2, 1) and CeO2 were 55.7, 49.5, 50.8, 36.6 and 22.7 m2 g–1, respectively. Surface area of ZrO2 is more than twice higher than that of CeO2. Interestingly the addition of small amounts of ZrO2 to CeO2 (about 20%) increases considerably the surface area of the catalyst. No dramatic change in the surface area of these catalysts is seen upon impregnation with 2 wt%. Rh. These surface areas were found equal to 46.7, 42.8, 43.6, 28.5 and 17.6 m2 g–1 for Rh/ZrO2, Rh/CeO2–ZrO2 (Ce/Zr = 4, 2, 1) and Rh/CeO2, respectively.
Fig. 1 shows TPR results of the different catalysts (supports and Rh-supported) as a function of hydrogen consumption per surface BET. ZrO2 incorporation into CeO2 has resulted in a higher reduction yield (per m2). Although some deviations occurred for the Rh-supported series, such a characteristic was qualitatively preserved for the supported Rh catalysts.
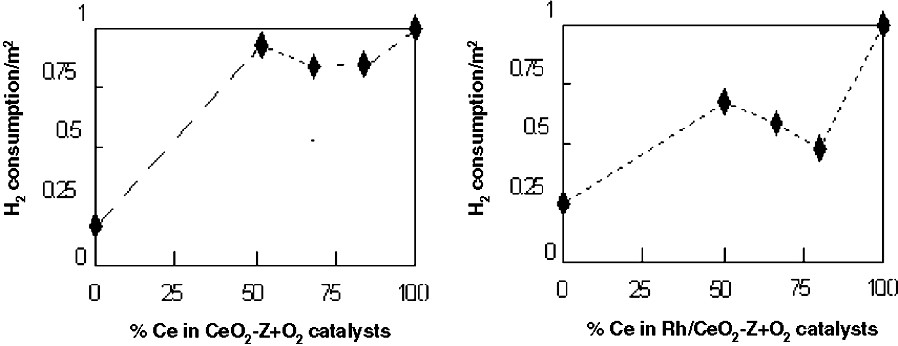
Hydrogen consumption per catalyst specific surface as a function of Ce content for: (a) Supports and (b) Rh catalyst.
Table 1 shows XPS results for all catalysts and compared to those of CeO2 and ZrO2 supports. In general there is no major deviation from the expected Rh atomic% on the surface when compared to the bulk (with the exception of the 2 wt% Rh/4 CeO2 – 1 ZrO2 catalyst). Some traces of Cl atoms (left from the preparation) are also found. For all catalysts, XPS Rh 3d lines were in an oxidised form (Rh3d5/2 at 309 eV (±0.2 eV depending on the catalyst). Although the difference in the binding energy between Rh3+ (Rh2O3) and Rh4+ (RhO2) is almost 1 eV, the small amount of Rh and the potential presence of ligand effect (due to proximity of Cl atoms) make the exact assignment rather difficult and not directly relevant for this study. XPS Ce 3d lines for all catalysts showed the presence of a small% of Ce3+ (except for CeO2 alone, where all Ce atoms where Ce4+). The spectra of XPS Ce 3d for oxidized and reduced surfaces have been published by us and other workers and can be found elsewhere [14–16]. Similar observation was seen for Zr 3d lines, where Zr2+ in addition to Zr4+ were also seen. The XPS Zr 3d lines of Rh/ZrO2 are very similar to those reported for ZrO2 containing materials that have been subject to particle bombardment [17,18]. When compared to CeO2 less efforts have been devoted to investigate the XPS Zr 3d lines for ZrO2–x (x < 1) in its reduced form. A representative spectrum for XPS Zr 3d lines for Rh/ZrO2 is given in Fig. 2 (and compared to that of stoichiometric ZrO2). We can also notice that the ratio Ce/Zr given for Rh/CeO2–ZrO2 catalyst is different from that expected from the bulk. If the ratio Ce/Zr is almost the same for CeO2–ZrO2 alone [19], literature shows that segregation occurs when a transition metal is present [20,21].
Atomic% of the surface and near surface elements of Rh catalysts and supports as determined by XPS.
| Catalyst | 2 wt% Rh/CeO2 | 2 wt% Rh/ZrO2 | 2 wt% Rh/CeO2–ZrO2, Ce/Zr a = | CeO2 | ZrO2 | ||
| 4 | 2 | 1 | |||||
| Ce | 26.8 | – | 26.5 | 34.1 | 19.6 | 23.5 | – |
| Zr | – | 30.9 | 8.9 | 12.2 | 6.2 | – | 23.7 |
| O | 70.2 | 66.4 | 62.1 | 50.2 | 71.8 | 76.5 | 76.3 |
| Rh | 3.0 (3.3) | 2.7 (2.4) | 2.5 (3.2) | 3.5 (3.1) | 2.4 (2.8) | – | – |
| Ce/Zr | 3.0 | 2.8 | 3.2 | ||||
| O/(Ce+Zr) | 2.6 | 2.2 | 1.78 | 1.1 | 2.8 | 3.3 | 3.2 |
a atomic ratios.
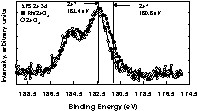
XPS Zr 3d lines for the as prepared Rh/ZrO2 and ZrO2.
We have previously investigated by XRD the bulk structure of the above catalysts [5]. CeO2 shows the expected diffraction of the fluorite structure while the monoclinic + tetragonal diffraction lines are shown for ZrO2. The absence of ZrO2 diffraction lines at low loading of ZrO2 into CeO2 (Ce/Zr = 4 and 2) indicates the presence of a solid solution. The shift of the diffraction lines to higher degrees is attributed to shrinking of the lattice due to the replacement of Ce ions (1.09 Å) by Zr ions (0.86 Å) [5,22]. At a higher loading of ZrO2 (Ce/Zr = 1), an additional tetragonal phase was also seen [5].
Table 2 (A and B) shows results from the catalytic reactions of the Rh catalysts as a function of temperatures. Both the conversion and product distributions are reported in Table 2A, while the yield of each product per mole of ethanol is given in Table 2B. With the exception of Rh/CeO2, all catalysts showed a 100% conversion at and above 300 °C. H2 production increases with increasing temperature, while that of CH4 declined. This may indicate that an additional formation of hydrogen occurs due to reforming of methane; methane is produced via the decomposition of acetaldehyde (see below Equations (4) and (5)). By 450 °C, all catalysts are virtually identical, reaching about 70 mol% production of H2 in selectivity at total conversion, while CH4 formation is below 5%. This composition is within experimental errors very close thermodynamic equilibrium: (H2 (70.64%); CH4 (3.4%); CO2 (22.1%); CO 53.8%)). This composition changes with the water/ethanol ratio [23].
Conversion (%) and product yield (%) at different temperatures for the Rh catalysts
| Rh/CeO2/Temperature (°C) | 300 | 350 | 400 | 450 |
| Conversion,% | 58.5 | 82.3 | 100 | 100 |
| Product Yield,% | ||||
| H2 | 59.7 | 64.7 | 66.3 | 69.1 |
| CH4 | 15.6 | 12.3 | 11.4 | 8.2 |
| CO | 18.2 | 5.4 | 3.7 | 3.5 |
| CO2 | 6.5 | 17.0 | 18.7 | 19.2 |
| CO2/CO | 0.4 | 3.1 | 5.1 | 5.5 |
| Rh/ZrO2 | ||||
| Conversion,% | 100 | 100 | 100 | 100 |
| Product Yield,% | ||||
| H2 | 59.6 | 63.5 | 68.1 | 71.7 |
| CH4 | 15.9 | 13.8 | 9.8 | 6.0 |
| CO | 13.1 | 4.3 | 1.6 | 2.1 |
| CO2 | 11.4 | 18.4 | 20.5 | 20.3 |
| CO2/CO | 0.9 | 4.3 | 12.8 | 9.4 |
| Rh/4 CeO2–1 ZrO2 | ||||
| Conversion,% | 100 | 100 | 100 | 100 |
| Product Yield,% | ||||
| H2 | 57.4 | 62.0 | 65.8 | 70.3 |
| CH4 | 18.2 | 15.4 | 12.1 | 7.2 |
| CO | 11.6 | 2.8 | 0.8 | 1.6 |
| CO2 | 12.7 | 19.7 | 21.3 | 20.8 |
| CO2/CO | 1.1 | 6.9 | 27.7 | 13.2 |
| Rh/2 CeO2–1 ZrO2 | ||||
| Conversion,% | 100 | 100 | 100 | 100 |
| Product Yield,% | ||||
| H2 | 56.9 | 58.7 | 64.2 | 69.2 |
| CH4 | 17.8 | 18.5 | 13.6 | 8.5 |
| CO | 16.9 | 4.7 | 1.8 | 1.6 |
| CO2 | 8.4 | 18. 2 | 20.3 | 20.7 |
| CO2/CO | 0.5 | 3.9 | 11.1 | 13.1 |
| Rh/1 CeO2–1 ZrO2 | ||||
| Conversion,% | 100 | 100 | 100 | 100 |
| Product Yield,% | ||||
| H2 | 57.1 | 59.6 | 64.4 | 70.3 |
| CH4 | 18.1 | 18.4 | 13.5 | 7.3 |
| CO | 13.5 | 0.8 | 0.7 | 1.5 |
| CO2 | 11.3 | 21.3 | 21.5 | 20.9 |
| CO2/CO | 0.8 | 28.0 | 32.6 | 14.0 |
Production in mole per mole of ethanol at different temperatures for the Rh catalysts
| Mole/mole of ethanol/Temperature (°C) | 300 | 350 | 400 | 450 |
| Rh/CeO2 | ||||
| H2 | 1.2 | 2.7 | 3.5 | 5.0 |
| CH4 | 0.3 | 0.5 | 0.6 | 0.6 |
| CO | 0.4 | 0.24 | 0.2 | 0.2 |
| CO2 | 0.1 | 0.7 | 1.0 | 1.4 |
| Rh/ZrO2 | ||||
| H2 | 2.9 | 3.8 | 4.9 | 5.7 |
| CH4 | 0.8 | 0.8 | 0.7 | 0.5 |
| CO | 0.6 | 0.3 | 0.1 | 0.2 |
| CO2 | 0.5 | 1.1 | 1.5 | 1.6 |
| Rh/4 CeO2–1 ZrO2 | ||||
| H2 | 2.5 | 3.7 | 4.3 | 5.5 |
| CH4 | 0.8 | 0.9 | 0.8 | 0.6 |
| CO | 0.5 | 0.2 | 0.1 | 0.1 |
| CO2 | 0.5 | 1.2 | 1.4 | 1.6 |
| Rh/2 CeO2–1 ZrO2 | ||||
| H2 | 2.8 | 3.5 | 4.1 | 5.3 |
| CH4 | 0.9 | 1.1 | 0.9 | 0.7 |
| CO | 0.7 | 0.1 | 0.1 | 0.1 |
| CO2 | 0.6 | 1.3 | 1.3 | 1.6 |
| Rh/1 CeO2–1 ZrO2 | ||||
| H2 | 2.6 | 3.2 | 4.4 | 5.8 |
| CH4 | 0.8 | 1.0 | 0.9 | 0.6 |
| CO | 0.6 | 0.2 | 0.1 | 0.1 |
| CO2 | 0.7.0 | 1.0 | 1.5 | 1.7 |
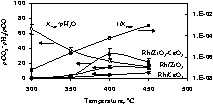
Contrary to hydrogen production, the CO2 to CO ratio changes dramatically from catalyst to catalyst. The lowest CO2/CO was seen for Rh/CeO2, while the most active catalysts are those containing both Zr and Ce oxides (Fig. 3).
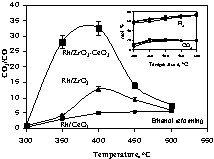
CO2/CO molar ratios for Rh/CeO2, Rh/ZrO2 and Rh/1 ZrO2–1 CeO2 catalysts as a function of reaction temperature; the inset shows both CO2 and H2 molar% for the same catalysts.
If we consider that at high temperatures reforming of methane by water is taking place followed by WGSR:
| (1) |
| (2) |
| (3) |
If we add to Equation (3) the following two equations:
| (4) |
| (5) |
Equation (2) is repeated twice because one CO is formed by Equation (5) and another by Equation (1).
Then, the total number of molecules of H2 per molecule of ethanol is equal to 6 (accompanied by two molecules of CO2). The inset in Fig. 3 shows the molar% of H2 and CO2 as a function of temperature for three representative catalysts (Rh/CeO2, Rh/ZrO2, and Rh/1 CeO2–1 ZrO2). The H2/CO2 molar ratio is very close to 3 at 450 °C and slightly over 3 at 500 °C (this falls within experimental uncertainties). In all cases, this shows that all catalysts are similarly active for the reforming of ethanol.
The high yield of CO2 on the series containing Zr might be tracked to the amount of available oxygen on the surface initially. Fig. 4 shows XPS O(1s) for the three Zr-containing catalysts and the CO2/CO molar ratio. The higher the surface oxygen the higher is the CO2/CO molar ratio. WGSR may operate in two ways [24]: (i) via a CO reaction with surface hydroxyls to make formate species that decompose to CO2 leaving water to dissociate on the oxygen vacancy (regeneration); (ii) CO is directly oxidised by surface oxygen to CO2 (Eley–Rideal mechanism) and again water dissociation regenerates the surface vacancy. Over CeO2 surfaces several workers have reported that regeneration of oxygen vacancies formed upon CO oxidation is due to water dissociation [25]. Both mechanisms involve an active surface oxygen transfer to oxidise CO to CO2. It is thus highly likely that for a similar material the higher the surface oxygen concentration, the higher is the possibility of CO oxidation.
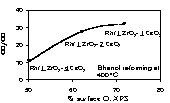
CO2/CO molar ratios as a function of surface percentage of O (1s) evaluated by XPS for Rh/1 ZrO2–1 CeO2, Rh/1 ZrO2–2 CeO2 and Rh/1 ZrO2–4 CeO2.
The decrease of CO2/CO molar ratio at high temperature (above 400°C) is attributed to thermodynamic limitations. WGSR is exothermic and is thus not favoured at very high temperature, while steam reforming is endothermic. The shape of the curves in Fig. 4 translates both reactions and the maximum is a compromise between them. Fig. 5 shows the computed
Experimental
As can be seen, within experimental errors at 400 °C and above, the formation of both H2 and CO2 is governed by the equilibrium constant for WGSR over the catalyst containing both Zr and Ce oxides (the most active catalyst). At higher temperatures, all catalysts behave similarly, because methane reforming has become very efficient, but the limitation is due to WGSR.
In summary, results of this work indicate the following two points: (1) the best catalyst for making hydrogen and CO2 is Rh supported over CeO2–ZrO2 and (2) the CO2/CO is sensitive to the Ce/Zr in the Rh/CeO2–ZrO2 series investigated and the production at high temperature reaches the thermodynamic equilibrium of WGSR.
Acknowledgements
The authors thank Peter Buchanan for his assistance during XPS data collection and Suzanne Libs for her help in gas-phase analyses of the catalytic reactions.