

1 Introduction
The N,N-dialkylcarbamato ligand is versatile, forming complexes with many elements within the Periodic Table [1]. N,N-dialkylcarbamato complexes have unique properties, and protic reagents [2] promptly displace coordinated carbon dioxide. The reactivity of N,N-dialkylcarbamato complexes has been extensively exploited in our laboratories for: (i) controlled hydrolysis in various M/H2O molar ratios [3]; (ii) exhaustive hydrolysis; (iii) implanting metal cations on inorganic surfaces, mainly silica [4]. The well-established hydrolysis experienced by homoleptic N,N-dialkylcarbamato metal complexes of formula M(O2CNR2)n, leads to μ-oxo derivatives (see Eq. (1)), the degree of oxide formation depending on the M/H2O ratio, and on the nature of M.
| (1) |
In apparent contrast with this reactivity, some metal oxides have been used in the past as synthetic precursors to N,N-dialkylcarbamato metal derivatives. Not only silver oxide [5], characterized by a low value, of the free energy of formation (Ag2O, ΔG0f = −11.2 kJ mol−1, i.e. −5.6 kJ mol−1 per mol of silver) [6], but also ZnO [7] (ΔG0f = −320.5 kJ mol−1 [6]) react according to Eqs. (2) and (3), respectively:
| (2) |
| (3) |
This paper reports some new results on the synthesis and characterization of new N,N-dialkylcarbamato complexes of magnesium, as obtained from the metal oxide. The synthesis of magnesium carbamato complexes has been recently reported by four different groups [8–11], the interest on this subject being justified by the probable presence of these systems in the active site of Rubisco [12], an enzyme involved in the carbon dioxide metabolism in living organisms. We therefore reckoned that the extension of the metal-oxide-based synthetic methodology to μn-O− or to homoleptic metal−dialkylcarbamato complexes of magnesium could represent an important acquisition in general terms and an even more challenging proposition in view of the higher stability of magnesium oxide (ΔG0f = −569.4 kJ mol−1) [6].
2 Results and discussion
The reaction of MgO with [Me2NH2][O2CNMe2] in toluene resulted in a relatively fast (1 d for the conversion of 2 g of MgO at room temperature) and substantially complete conversion to products of formula [Me2NH2]n[Mg8(CO3)2(O2CNMe2)12+n], with n ranging from 3 to 0 (see Eq. (4)).
| (4) |
Product composition was found to depend on work-up conditions, especially temperature and time during the recovery, the solid being treated in vacuo before analysis. Products of the two limiting compositions, [Me2NH2]3[Mg8(CO3)2(O2CNMe2)15], 1, and Mg8(CO3)2(O2CNMe2)12, 2, were obtained by appropriate work−up, see Experimental Section. The latter compound was obtained from 1 by gradual releasing of [Me2NH2][O2CNMe2], as confirmed by the weight loss experienced by a sample of 1 when heated in vacuo. The CO2/Mg molar ratio varies consistently from 2.06 to 1.74, the theoretical values being 2.12 and 1.75 for 1 and 2, respectively.
| (5) |
| (6) |
| (7) |
We propose the presence in solution of equilibria (5)−(7). This is substantiated by the observation that 2 has been converted back into 1 by treatment in toluene with an excess of dimethylammonium N,N-dimethylcarbamate under a carbon dioxide atmosphere. Addition of heptane to the toluene solution of the primary products in the presence of [Me2NH2][O2CNMe2] will precipitate the ionic products (1 was recrystallized at 4 °C in a satisfactory yield), while removing the volatiles in vacuo at 60 °C will form 2, that is sparingly soluble in organic solvents. A similar release of [R2NH2][O2CNR2] was observed for [NH2iPr2][Ti2(O2CNiPr2)7] giving Ti2(O2CNiPr2)6 [13].
The analysis of the IR and NMR spectra of 1 is complicated by the high number of non-equivalent carbamato ligands. A broad strong band is present in the nujol IR spectrum in the range 1640−1450 cm−1, attributable to a number of slightly different C−O and C−N stretching vibrations of the carbamato and carbonato ligands, suggesting various coordination modes. Carbamato [1] and carbonato [14] derivatives are reported to show stretching vibrations in the range 1680–1450 cm−1.
In the 1H−NMR spectra of product 1, signals due to carbamato methyl groups coalesce in a broad band centred at 2.8 ppm, while a broad signal at about 2.4 ppm is due to the ammonium methyl groups. Broad 13C−NMR signals are also present for the methyl carbons (36.2 ppm) and for the carbamato/carbonato CO2 moieties at 162.8 ppm. As the signals are rather broad, it is reasonable that those due to the CO2 moieties of the carbonato and carbamato groups overlap. They are in fact expected to be quite close: for instance in Mg5(CO3)(O2CNiPr2)8(HMPA)2 [11], the resonances are at 162.49 and 163.85 ppm for the carbamato and carbonato groups, respectively. Signals of low intensity attributable to crystallization solvents (toluene and heptane) are present even in samples extensively dried in vacuo at room temperature. Unfortunately, the low solubility of 2 prevents its NMR spectra to be recorded.
As already observed in the case of ZnO [7], dimethylamine is unique in reacting with MgO under absorption of CO2 to give the carbamato complexes. As a matter of fact, no products were obtained with NHEt2, NHiPr2 and NHBu2.
Attempts have also been made to obtain N,N-dimethylcarbamato complexes starting from magnesium metal and [NH2Me2][O2CNMe2]. As magnesium has a low standard reduction potential, this appeared to be a practicable route, similar to what already established for sodium [15]. A preliminary unsuccessful attempt was carried out between magnesium and NHiPr2/CO2 [8]. However, since we had already shown that the reactivity of the NHMe2/CO2 system, as compared with other NHR2/CO2 systems, is somehow special [7], this unsuccessful first attempt did not discourage us from trying with dimethylamine. We have found that magnesium reacts quite slowly with NHMe2/CO2 in toluene with di-hydrogen evolution affording 1, albeit in low yields. Thus, we assume that the direct reaction between Mg and [NH2Me2][O2CNMe2] is exceedingly slow and MgO impurities on the surface of the metal and accidental presence of water may promote the process. In fact, yields increase when commercial magnesium turnings (99.0%) or commercial [NH2Me2][O2CNMe2] are used instead of high-purity magnesium (99.9%) or freshly prepared [NH2Me2][O2CNMe2].
The slow and incomplete reaction of magnesium with [NH2Me2][O2CNMe2] agrees with the previous observation [16] that zinc of the highest available purity (99.999%) fails to react with the NHMe2/CO2 system, while zinc of a lower purity led to the corresponding homoleptic carbamato derivative of zinc(II) [NH2Me2][Zn2(O2CNMe2)5]·CH3CN.
Product 1, as recrystallized from hot heptane under a carbon dioxide atmosphere, was studied by single crystal X-ray diffraction methods. In the octanuclear aggregate, containing lattice heptane, [Me2NH2]3[Mg8(CO3)2(O2CNMe2)15]·C7H16, each magnesium atom is hexacoordinated in an octahedral geometry to the oxygen atoms of the carbamato and carbonato groups. A similar octahedral MgO6 core is encountered in the Rubisco enzymes, whose catalytic activity towards carboxylation is believed to rely on the formation of a magnesium carbamato complex resulting from the interaction of CO2 with an amino group of lysine at the active site of the enzyme [12].
Fig. 1 displays a view of the octanuclear ionic aggregates [Me2NH2]3[Mg8(CO3)2(O2CNMe2)15] of 1, while the Mg−O bond distances, shown in the figure as filled sticks, are reported in Table 1. The anion contains a ring of eight edge- or apex-sharing {MgO6} distorted octahedra, where the Mg–O distances span a rather large range of values from 1.921 to 2.313 Å. Also the O–Mg−O angles are rather far from the ideal octahedral values, e.g. 58.2° for O1−Mg2−O2 and 174.8° for O111−Mg5−O3. The framework is kept together by two μ6 carbonato ligands and by the fifteen carbamato ligands. The latter adopt four different ligation types. Scheme 1 shows the four types arranged in the decreasing order of frequency.
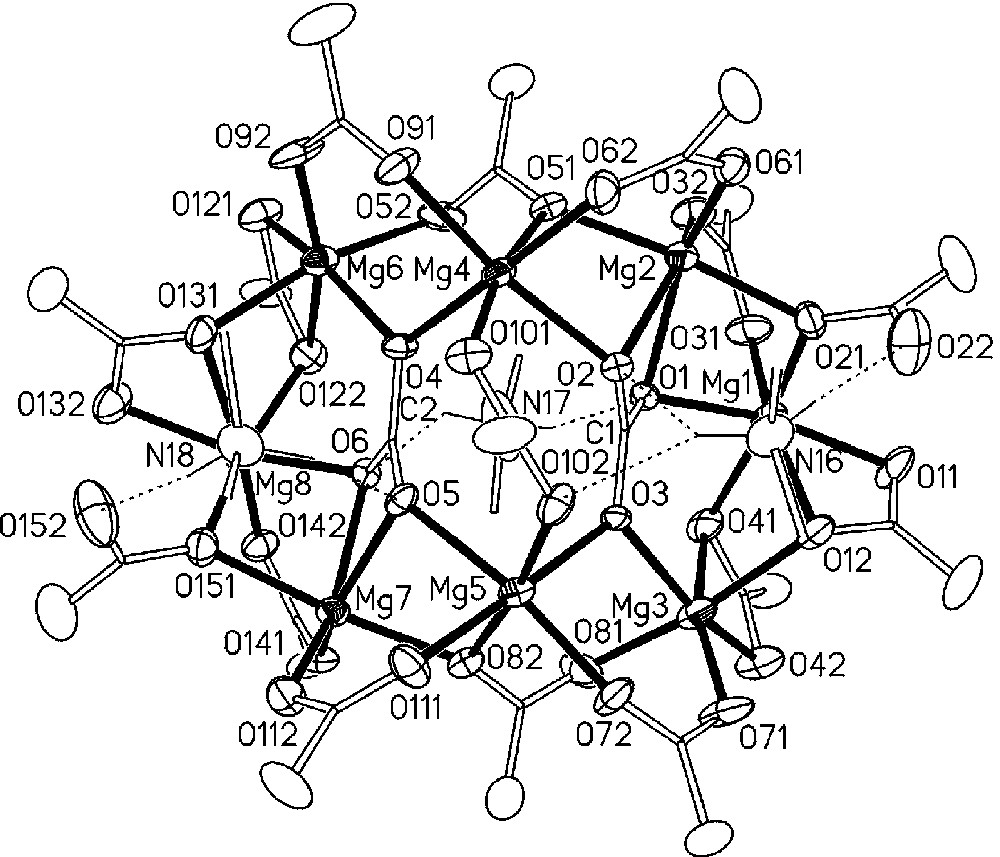
View of the structure of the ionic [NH2Me2]3 [Mg8(CO3)2(OCONMe2)15] heptane, 1. The thermal ellipsoids of the C atoms have been omitted for clarity, those of the Mg, O and N atoms have been represented at 30% probability. The dotted lines represent the main H-interactions.
Co-ordination distances (Å) of Mg in the structure of [Me2NH2]3 [Mg8(CO3)2(O2CNMe2)15]·heptane, 1
| Mg1−O31 | 1.945(10) | Mg5−O72 | 1.921(9) |
| Mg1−O11 | 1.996(10) | Mg5−O102 | 2.021(10) |
| Mg1−O1 | 2.068(9) | Mg5−O111 | 2.043(10) |
| Mg1−O41 | 2.093(10) | Mg5−O5 | 2.104(9) |
| Mg1−O21 | 2.070(10) | Mg5−O82 | 2.108(9) |
| Mg1−O12 | 2.251(9) | Mg5−O3 | 2.195(9) |
| Mg2−O32 | 1.967(10) | Mg6−O92 | 1.944(10) |
| Mg2−O61 | 2.007(10) | Mg6−O52 | 2.045(11) |
| Mg2−O51 | 2.031(9) | Mg6−O131 | 2.056(10) |
| Mg2−O21 | 2.046(10) | Mg6−O121 | 2.102(10) |
| Mg2−O1 | 2.187(9) | Mg6−O4 | 2.139(9) |
| Mg2−O2 | 2.252(9) | Mg6−O122 | 2.300(10) |
| Mg3−O71 | 1.963(11) | Mg7−O112 | 1.979(10) |
| Mg3−O81 | 1.990(11) | Mg7−O141 | 1.985(9) |
| Mg3−O42 | 2.070(10) | Mg7−O82 | 2.025(9) |
| Mg3−O12 | 2.107(11) | Mg7−O151 | 2.082(10) |
| Mg3−O3 | 2.177(9) | Mg7−O6 | 2.179(9) |
| Mg3−O41 | 2.313(10) | Mg7−O5 | 2.244(9) |
| Mg4−O91 | 1.983(10) | Mg8−O142 | 1.973(10) |
| Mg4−O62 | 1.989(10) | Mg8−O6 | 2.030(9) |
| Mg4−O101 | 2.030(10) | Mg8−O132 | 2.032(10) |
| Mg4−O2 | 2.128(9) | Mg8−O122 | 2.044(10) |
| Mg4−O51 | 2.153(9) | Mg8−O151 | 2.072(10) |
| Mg4−O4 | 2.169(9) | Mg8−O131 | 2.248(10) |
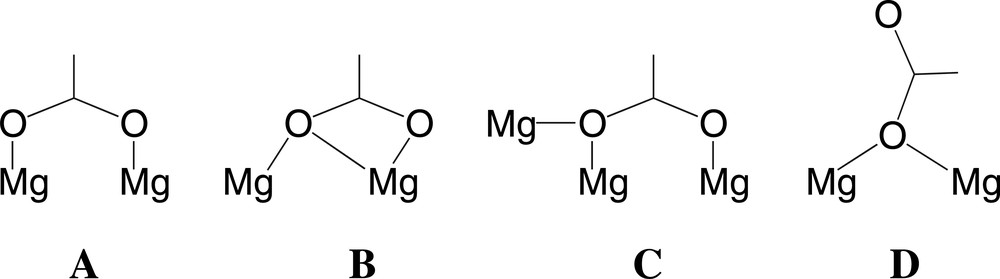
While ligation types A, B and C have been encountered previously [1], the bridging type D has never been observed before. It is interesting to note that the Mg−O bond distances longer than 2.30 Å are related to the carbamato groups 4 and 12, both adopting the ligation type B. The octanuclear aggregate is completed by three dimethylammonium cations, whose nitrogen atoms are labelled N16, N17 and N18, which are disposed around the anion through both ionic bonds and a number of N−H···O interactions shown in the figure by dotted lines. The parameters of the main H-interactions are listed in Table 2.
Main H-interactions in the ion quartet [Me2NH2]3 [Mg8(CO3)2(O2CNMe2)15]·heptane
| D−H···A | d(D−H) | d(H···A) | d(D···A) | <(DHA) |
| N16−H16A···O2 | 0.90 | 2.08 | 2.858(13) | 143.9 |
| N16−H16A···O102 | 0.90 | 2.36 | 3.018(14) | 130.3 |
| N16−H16B···O22 | 0.90 | 1.80 | 2.671(15) | 161.3 |
| N17−H17A···O6 | 0.90 | 2.17 | 2.907(14) | 139.0 |
| N17−H17B···O1 | 0.90 | 2.01 | 2.886(14) | 163.6 |
| N18−H18A···O152 | 0.90 | 1.74 | 2.631(14) | 168.9 |
| N18−H18B···O5 | 0.90 | 2.06 | 2.900(13) | 155.3 |
As it can be seen in both Figs. 1 and 2, the dimethylammonium cation containing the N16 nitrogen atom presents a multiple interaction through one of its hydrogen atoms. Moreover, the carbamato ligation of type D (see Scheme 1), which is not found in homoleptic metal carbamates [1]. is present in 1, because the oxygens O22 and O152 accept the strongest hydrogen bonds.
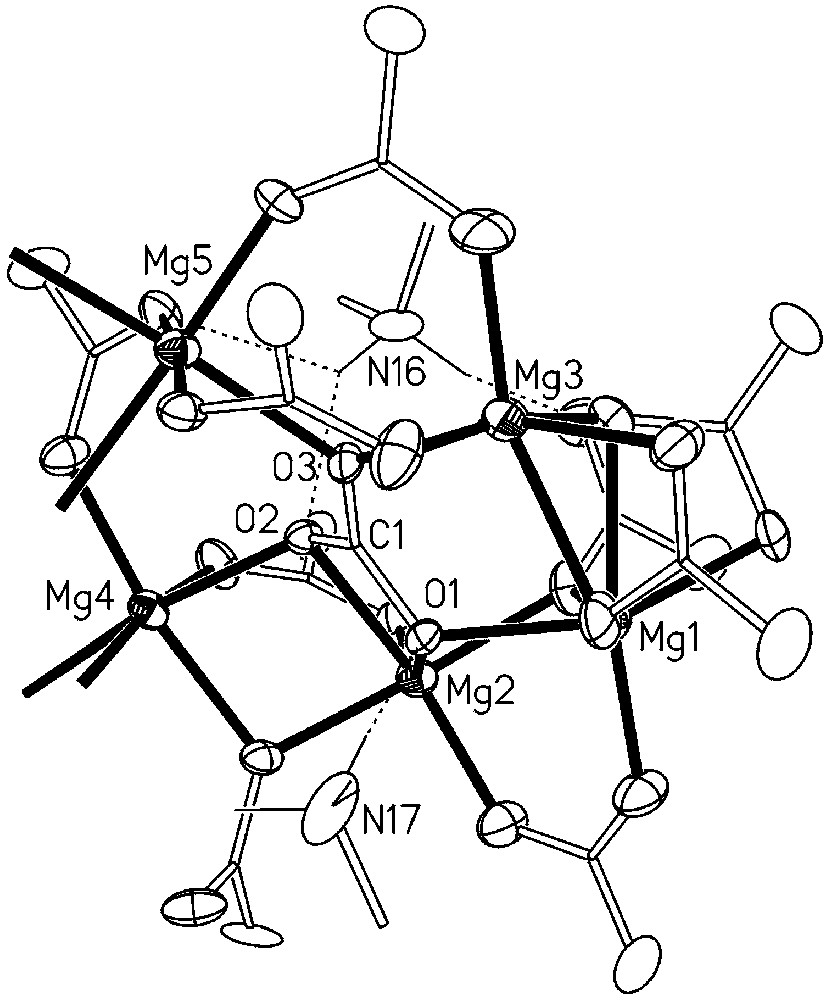
Detailed view of the coordination of the carbonato ligand in 1 (C1, O1, O2, O3).
The octanuclear aggregate has approximate C2 symmetry, with the twofold axis passing through N17 and the carbamato group 10. This feature is easily recognized in Fig. 1, which is projected almost exactly in this direction. The octanuclear ionic aggregates [NH2Me2]3[Mg8(CO3)2(OCONMe2)15] are packed in the crystal alternated with heptane molecules, and only van der Waals interactions are present between adjacent octanuclear aggregates. The strong ionic and hydrogen interactions that connect the components of each aggregate probably survive in solvents of low polarity, which explains the solubility of this substance in toluene.
Fig. 2 shows the coordination of the carbonato group containing the carbon atom labelled C1. We have taken advantage of the presence of the pseudo C2 symmetry to simplify the drawing and to represent only one half of the aggregate, the other half being almost equivalent. As shown in Fig. 2, the O3 atom of the substantially planar carbonato ligand is coordinated to C1, Mg3 and Mg5, being only 0.009 Å out of the plane defined by these three atoms. On the contrary, the oxygen atoms O1 and O2 are co-ordinated to C1, Mg1, Mg2 and to C1, Mg2, Mg4, respectively, with a marked pyramidal geometry, being 0.691 and 0.577 Å, respectively, out of the coordination planes. These deviations from planarity can be ascribed to the acceptance of strong hydrogen bonds from the ammonium ions containing N17 and N16, respectively.
The architecture of the aggregate with the two μ6-carbonato ligands resembles that of [Mg9(μ5-CO3)(O2COMe)8(μ3−OMe)8(MeOH)13]· MeOH · C7H8, as reported by Mingos et al. [17]. In this case, the presence of the carbonato groups has been explained as due to partial hydrolysis of the precursor [Mg(O2COMe)(OMe)(MeOH)1.5]n. In the ennea–nuclear complex, the Mg−O distances within the μ5-CO3 ligand range between 2.031 and 2.148 Å. Recently the synthesis of a magnesium carbamato−carbonato complex Mg5(μ5-CO3)(O2CNiPr2)8(HMPA)2 has been reported [11], where the magnesium atoms form a five-membered ring through the bridging carbonato group.
The μ5 coordination hapticity of a carbonato ligand is unusual. Even more rare is the μ6 coordination mode of the carbonato ligand. A literature survey has pointed out the existence of a few cases of this coordination mode for vanadium [18], molybdenum [19,20], nickel [21], and lanthanides [22].
3 Experimental section
All preparations were carried out in standard Schlenk tubes. All solvents were freshly distilled over conventional drying agents under dinitrogen and all reactions were carried out under an atmosphere of dinitrogen, or carbon dioxide, as indicated. Dimethylammonium dimethylcarbamate, [Me2NH2][O2CNMe2], was purchased from Fluka or freshly prepared from dimethylamine and carbon dioxide, as specified. Magnesium oxide (99.9%) was obtained by calcining commercial MgH2 (Aldrich) and kept in sealed vials under a dry atmosphere. Elemental analysis: found% (calc.% for MgO): Mg 59.4 (60.3). The metal content of commercial magnesium in turnings (Aldrich) was 99.0%. Pure magnesium metal (≥ 99.9%) was obtained as the unreacted residue (used in excess) after treatment of commercial magnesium with [Me2NH2][O2CNMe2].
IR spectra were measured with a Perkin Elmer FT−IR mod. 1725X spectrophotometer. NMR spectra were recorded using a Varian Gemini 200 MHz instrument, the data being expressed in ppm from TMS for 1H and 13C. GC analyses were carried out on a Dani 3800 gas chromatograph. Elemental analyses (C, H, N) were performed by the ‘Laboratorio di Microanalisi, Facoltà di Farmacia, Università di Pisa’, with a Carlo Erba mod. 1106 elemental analyzer. Magnesium analyses were carried out by complexometry [23] using standard EDTA solutions. Carbon dioxide was determined by gas-volumetric measurements, the samples being attacked with 20% sulphuric acid.
3.1 Reaction of MgO with [Me2NH2][O2CNMe2]
To a toluene (100 ml) solution of [Me2NH2][O2CNMe2] (32.0 ml, 0.250 mol) freshly prepared from NHMe2 and CO2, MgO (1.32 g, 32.76 mmol) was added under an atmosphere of carbon dioxide. After 1 d stirring at room temperature, the mixture was filtered under carbon dioxide to remove traces of unreacted magnesium oxide. By removing all the volatiles from the filtrate at room temperature under reduced pressure, a colourless residue was obtained. Upon addition of heptane (100 ml), the product was recovered by filtration of the resulting suspension under dinitrogen. The solid was dried in vacuo (5.21 g, 84% yield based on the starting magnesium). Elemental analysis: found% (calc.% for [NH2Me2][Mg8(CO3)2(O2CNMe2)13], C43H86N14Mg8O32): Mg 12.8 (12.9), CO2 43.9 (43.8), corresponding to a CO2/Mg molar ratio of 1.89 (th. 1.87)). IR (nujol, 3000−600 cm−1): 1605 (sh), 1567 (s), 1507 (s), 1282 (m). The analytical data were found to depend on the drying procedure: variable compositions, [Me2NH2]n[Mg8(CO3)2(O2CNMe2)12+n], with n ranging from 3 to 0, were obtained. Similar results were obtained by using commercial [Me2NH2][O2CNMe2].
Starting from commercial MgO (Aldrich, fused, pieces, 99.96%), the reaction was found to be extremely slow at room temperature.
3.2 Reaction of Mg with [Me2NH2][O2CNMe2]
Freshly prepared (T ~ −10 °C) [Me2NH2][O2CNMe2] from dimethylamine (30 ml, 0.65 mol) and carbon dioxide was reacted with commercial magnesium turnings (3.00 g, 0.12 mol) in toluene (100 ml). The suspension was stirred and a slow evolution of di-hydrogen was confirmed by a gas-chromatographic control. Vacuum/carbon dioxide cycles were repeated at regular intervals of time. After 12 d, the unreacted metal was filtered off, and the solution was evaporated to dryness in vacuo at room temperature. The solid residue was suspended in heptane, filtered under di-nitrogen and dried in vacuo. The colourless product was recovered (1.21 g, 5% yield based on the starting magnesium). Elemental analysis: found% (calc.% for [Me2NH2][Mg8(CO3)2(O2CNMe2)13], C43H86N14Mg8O32): Mg 13.1 (12.9), CO2 41.7 (43.8), corresponding to a CO2/Mg molar ratio of 1.76 (th. 1.87)). IR (nujol, 2000-1250 cm–1): 1611 (sh), 1568 (sh), 1515 (s), 1283 (s). 1H–NMR (C6D6, δ): 2,9 (m, 81H); 2,3 (s, 19H) ppm.
The reaction was also carried out starting from commercial [Me2NH2][O2CNMe2]. After 13 d, the unreacted metal was filtered off and the solution was evaporated to dryness in vacuo at room temperature. The work-up was as in the previous syntheses, taking care in this case to keep the temperature below 30 °C throughout the whole procedure (39.4% yield with respect to magnesium metal). Elemental analysis: found% (calc.% for [Me2NH2]3[Mg8(CO3)2(O2CNMe2)15], C53H114Mg8 N18O36: Mg 11.4 (11.0), CO2 42.6 (42.2), corresponding to a CO2/Mg molar ratio of 2.06 (2.12)). IR (nujol, 2000−1250 cm−1): 1611 (sh), 1582 (sh), 1515 (s), 1283 (s). 1H−NMR (C6D6, δ): [2.9; 2.8] (81 H); 2.4 (19 H) ppm. 13C–NMR (C6D6, δ): 162.8; 36.2 ppm. This compound was recrystallized from hot heptane under carbon dioxide and single crystals suitable for X-ray diffraction studies were thus obtained.
When high-purity (99.9%) magnesium was treated with freshly prepared [Me2NH2][O2CNMe2], after 1 month, about 80% of the starting magnesium was recovered by filtration and only a negligible amount of a solid residue was obtained by removing all the volatiles from the filtrate.
3.3 Crystallographic study
Crystals of 1 were small and poorly diffracting. A crystal of dimensions 0.32 × 0.28 × 0.18 mm was selected under a dinitrogen atmosphere saturated with heptane and sealed in a glass capillary. The diffractions were collected at room temperature with a Bruker SMART CCD area detector diffractometer. Crystal data are reported in Table 3.
Crystal data and structure refinement
| Empirical formula | C60H130Mg8N18O36 |
| Formula weight | 1874.30 |
| Temperature (K) | 293(2) |
| Crystal system | Monoclinic |
| Space group | P21/n (No. 14) |
| a (Å) | 14.305(1) |
| b (Å) | 37.079(3) |
| c (Å) | 18.940(2) |
| β (°) | 92.215(2) |
| U (Å3) | 10038(2) |
| Z | 4 |
| Dcalc (Mg m−3) | 1.240 |
| μ (mm−1) | 0.144 |
| Data/restr./param. | 13990/0/885 |
| R(Fo) [I > 2 σ(I)] a | 0.1034 |
| Rw (Fo2) [I > 2 σ(I)] a | 0.2233 |
| Goodness of fit a on F2 | 0.695 |
a R(Fo) = Σ∣∣Fo∣−∣Fc∣∣/Σ∣Fo∣; Rw(Fo2) = [Σ[w(Fo2 – Fc2)2]/Σ[w(Fo2)2]]1/2; w = 1/[σ2 (Fo2) + (A Q)2 + B Q], where Q = [MAX(Fo2,0) + 2 Fc2]/3;
Graphite-monochromatized Mo–Kα (λ = 0.71073 Å) radiation was used with the generator working at 50 kV and 35 mA. Cell parameters and orientation matrix were obtained from least-squares refinement on 315 reflections measured in three different sets of 15 frames each, in the range 0 ≤ θ ≤ 23°. The diffraction pattern showed measurable intensities only at low θ angle, so we limited the data collection to θ ≤ 23° in a half sphere (scan method), with the sample–detector distance being fixed at 5 cm. Only 20% of the measured reflections had I > 2 σ(I). No crystal decay was observed, and absorption correction was not applied owing to the relatively low absorption factor. A total of 27701 reflections were collected (13990 unique, Rint = 0.159; Rint = [Σ∣Fo2 – Fo2(mean)∣/Σ Fo2]. The structure was solved by direct methods (SIR-92) [24] and refined with full-matrix-block least-squares (SHELX-97) [25]. Anisotropic temperature factors were assigned to all non-hydrogen atoms. Hydrogens were let ride on their carbon or nitrogen atoms.
3.4 Interconversion [NH2Me2]3[Mg8(CO3)2(O2CNMe2)15] ÷ Mg8(CO3)2(O2CNMe2)12
A toluene (60 ml) solution of [NH2Me2]3[Mg8(CO3)2(O2CNMe2)15] (2.18 g, 1.23 mmol) was evaporated to dryness in vacuo at 50 °C. The residue (1.68 g, 84% yield) was sealed in vials. Elemental analysis: found% (calc.% for Mg8(CO3)2(O2CNMe2)12, C38H72N12Mg8O30]: Mg 13.7 (14.2), CO2 43.2 (44.9), corresponding to a CO2/Mg molar ratio of 1.74 (th. 1.75)).
The magnesium complex Mg8(CO3)2(O2CNMe2)12 (2.05 g, 1.49 mmol was dissolved in toluene (50 ml) in the presence of [Me2NH2][O2CNMe2] (5.0 ml, 0.04 mmol). The product was recovered as colourless crystals upon cooling the resulting solution at 4 °C. The product was dried in vacuo at a temperature below 30 °C (2.53 g, 96% yield). Elemental analysis: found% (calc.% for [NH2Me2]3[Mg8(CO3)2(O2CNMe2)15], C53H114N18Mg8O36: Mg 10.7 (11.0)).
Acknowledgments
This work was supported by the ‘Ministero dell’ Istruzione, dell' Università e della Ricerca', MIUR, ‘Progetti di Rilevante Interesse Nazionale’ 2002–2003 and by the ‘Consiglio Nazionale delle Ricerche’, ‘Progamma CNR/MIUR Nanotecnologic, Processi litografici per nanofabbricazione’. The authors wish to thank Professor L. Busetto of the University of Bologna for allowing us to collect the X-ray data on their Bruker SMART CCD area detector diffractometer and Dr. E. Di Martino for help during data collection.
Supplementary material
Details about the structure determination have been deposited in the form of the CIF file with the Cambridge Crystallographic Data Centre as deposit number CCDC 225857 for 1. Copies of the data can be obtained, free of charge, on application to the CCDC, 12 Union Rd, Cambridge CB21EZ UK (fax: +44 –1223-336-033; e-mail: deposit@ccdc.cam.ac.uk).