

1 Introduction
In the course of our work on redox active phosphines and diphosphines bearing the tetrathiafulvalenyl core, we have described the synthesis of tertiary phosphines incorporating one, two or three TTF moieties as in (TTF)PPh2 [1], (TTF)2PPh [1] and (TTF)3P [2]. These preparations are based on the ability of the hydrogen atoms of the TTF core to be abstracted by strong bases such as LDA to form the corresponding lithium derivatives [3]. Reaction with a variety of electrophiles is then possible, as for example chlorophosphines. Substituted TTFs like o-Me2TTF and Me3TTF were also functionalized through the same procedure, that is, lithiation and reaction with ClPPh2, Cl2PPh or Br3P to afford respectively the mono-(o-Me2TTF)PPh2 and (Me3TTF)PPh2, bis-(o-Me2TTF)2PPh and (Me3TTF)2PPh, and tris-(o-Me2TTF)3P and (Me3TTF)3P tetrathiafulvalenyl phosphines [4,5]. Those phosphines incorporating two or three TTF redox moieties are particularly interesting to determine the degree of delocalisation of radical species obtained upon oxidation, as investigated by extensive EPR-ENDOR studies [5] (Scheme 1).
Also, we were attracted by the possibility offered by the tris(TTF)phosphines such as (TTF)3P, (o-Me2TTF)3P and (Me3TTF)3P to organize in the solid state, in their neutral or oxidized form, into a three-fold symmetry structural organization that would reflect the local symmetry around the phosphorus atom. In that respect, among the nine phosphines described above, only (TTF)3P has been structurally characterized [2], and it crystallizes in general position in the monoclinic system, space group C2/c. We have therefore undertaken the structure resolution of the three phosphines derived from o-Me2TTF, that is the mono- (o-Me2TTF)PPh2 (1), bis- (o-Me2TTF)2PPh (2), and tris- (o-Me2TTF)3P (3) phosphines, with special attention to the latter.
2 Experimental
2.1 Syntheses
The three redox active phosphines 1−3 were prepared as previously described [4,5] from o-Me2TTF-Li and ClPPh2 for 1, 2 equiv o-Me2TTF-Li and Cl2PPh for 2 and 3 equiv o-Me2TTF-Li and PBr3 for 3. Recrystallization of 1 from pentane, 2 from toluene and 3 from chlorobenzene afforded suitable crystals for X-ray structure determination.
2.2 Crystallographic data collection and structure determination
Single crystals were mounted on a STOE Imaging Plate Diffractometer (IPDS) with a graphite-monochromated Mo Kα radiation source (λ = 0.71073 Å). Data were collected at 293(2) K. Structures were solved by direct methods (SHELXS-97) and refined with full-matrix least-squares method on F2 using SHELXL-97 programs. Hydrogen atoms were introduced at calculated positions and not refined (riding model). Crystallographic data are given in Table 1. Full-bond lengths and bond angles, atomic coordinates and complete crystal structure results are deposited as supplementary materials in cif format.
Crystal data and structure refinement
| (o-Me2TTF)PPh2 (1) | (o-Me2TTF)2PPh (2) | (o-Me2TTF)3P (3) | |
| Empirical formula | C20H17PS4 | C22H19PS8 | C24H21PS12 |
| Formula weight | 416.55 | 570.82 | 725.22 |
| Crystal system | monoclinic | monoclinic | trigonal |
| Space group | P21/c | P21/c | R3c |
| a (Å) | 14.3732(9) | 14.4560(13) | 21.285(2) |
| b (Å) | 6.0120(3) | 8.6624(5) | 21.285(2) |
| c (Å) | 23.1582(14) | 20.7713(19) | 12.0797(9) |
| α (°) | 90.0 | 90.0 | 90.0 |
| β (°) | 95.414(7) | 102.230(11) | 90.0 |
| γ (°) | 90.0 | 90.0 | 120.0 |
| V (Å3) | 1992.2(2) | 2542.0(4) | 4739.3(7) |
| Z | 4 | 4 | 6 |
| dcalc (g cm−3) | 1.389 | 1.492 | 1.524 |
| μ (mm−1) | 0.558 | 0.776 | 0.897 |
| Reflections collected | 19953 | 20245 | 15851 |
| Independent reflections | 3813 | 4648 | 2070 |
| [I > 2 σ(I)] | 2592 | 1441 | 1047 |
| Final R1a, wR2b | 0.0336, 0.0807 | 0.0421, 0.0833 | 0.0245, 0.0499 |
| Res. density (e− Å−3) | +0.30, –0.20 | +0.22, –0.23 | +0.20, –0.25 |
a R1 = Σ||Fo| − |Fc||/Σ|Fo|
b wR2 = [Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]]1/2
3 Results and discussion
3.1 Structure of (o-Me2TTF)PPh2 (1)
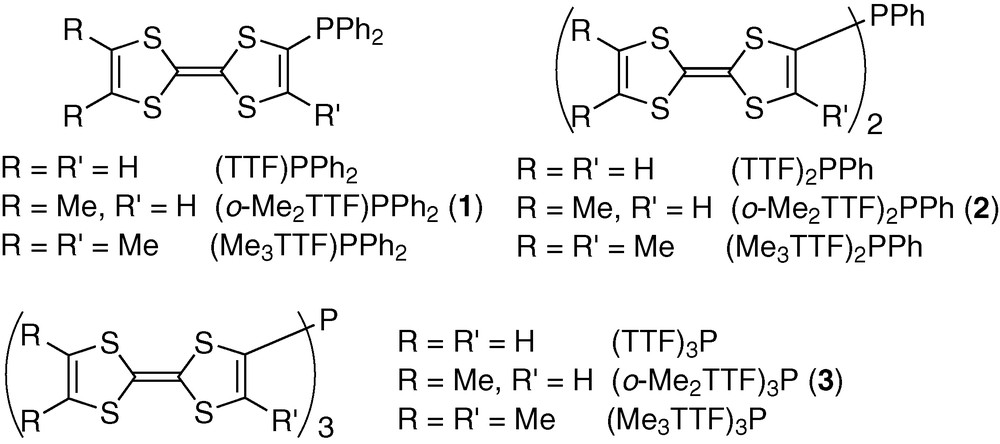
(o-Me2TTF)PPh2 crystallizes in the monoclinic system, space group P21/c with one molecule in general position in the unit cell (Fig. 1). Bond distances and angles are in the expected range, selected values are given in Table 2 and compare well with reference data for TTF [6] and o-Me2TTF [7]. The two dithiole rings of the TTF moiety are essentially planar, with folding angles along the S---S hinges of 3.2(1) and 3.3(1)° for the dimethyldithiole and the bis(diphenylphosphino)dithiole respectively. The phosphines organize into columns with the phosphorus lone pair pointing toward the same direction within a column and the TTF moieties stacking on top of each other along the b-axis with a plane-to-plane distance of 3.72 Å (Fig. 2). Note that no TTF···TTF dyad formation is found, while such a dyadic motif is very often observed in the solid-state structure of neutral TTF molecules [8].

View of the unit cell of (o-MeTTF)PPh2 (1). Phosphorus atoms are in dark grey, sulphur atoms in light grey.
Selected bond distances (Å) and angles (°) for 1−3 and reference compounds TTF, o-Me2TTF and (TTF)3P. The C=C distances refer to the central double bond of the TTF moieties, the C−S distances to those bonds involving carbon atoms of the central C=C group
| Compound | P−C | C−P−C | C=Ccentral | Ccentral−S a | ref. |
| TTF | — | — | 1.349(3) | 1.756(2) | [6] |
| o-Me2TTF | — | — | 1.340(4) | 1.753(4) | [7] |
| (o-Me2TTF)PPh2 (1) | 1.817(2) (TTF) | 104.1(1) | 1.330(3) | 1.759(4) | this work |
| 1.836(2) (Ph) | 101.9(1) | ||||
| 1.842(2) (Ph) | 100.5(1) | ||||
| (o-Me2TTF)2PPh (2) | 1.801(5) (A) | 103.1(2) | 1.350(6) (A) | 1.747(5) (A) | this work |
| 1.808(5) (B) | 100.1(2) | 1.341(6) (B) | 1.751(5) (B) | ||
| 1.814(6) (Ph) | 105.3(3) | ||||
| (o-Me2TTF)3P (3) | 1.815(4) | 102.7(1) | 1.345(4) | 1.756(5) | this work |
| (TTF)3P | 1.839(4) (A) | 101.4(2) | 1.318(6) (A) | 1.762(4) (A) | [2] |
| 1.806(5) (B) | 100.8(2) | 1.358(8) (B) | 1.748(8) (B) | ||
| 1.807(4) (C) | 103.5(2) | 1.338(5) (C) | 1.754(6) (C) |
a the four Ccentral−S distances have been averaged in each independent TTF moiety.
Stacking of the (o-MeTTF)PPh2 molecules along the b-axis.
3.2 Structure of (o-Me2TTF)2PPh (2)
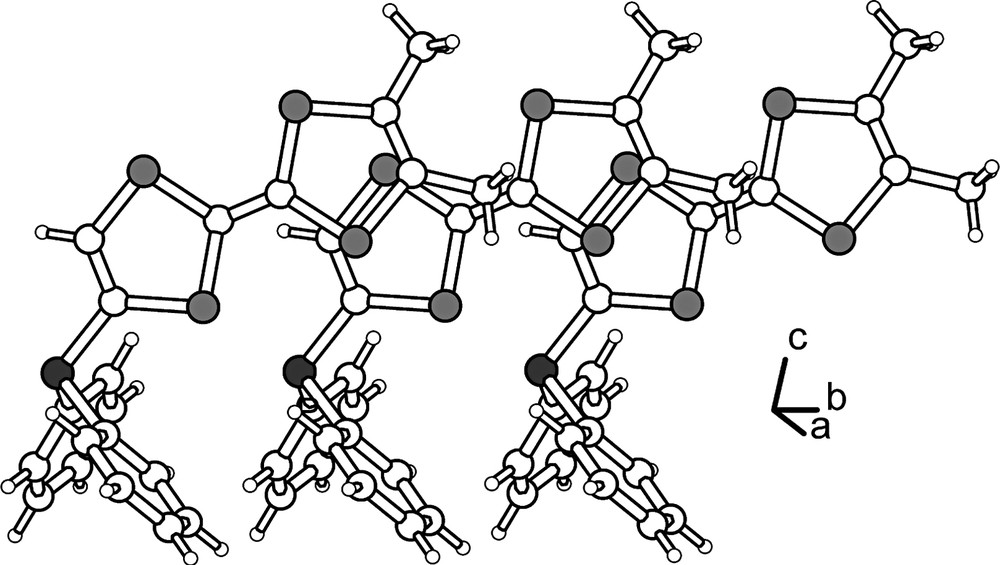
(o-Me2TTF)2PPh crystallizes in the monoclinic system, space group P21/c with one molecule in general position in the unit cell (Fig. 3). Bond distances and angles are in the expected range, and selected values are collected in Table 2. While the dimethyldithiole ring is still essentially planar, with a folding angle along the S---S hinge of 3.8(2) and 3.6(2)° in TTF A and B respectively, the dithiole rings linked to the phosphorus atom exhibit a sizeable distortion from planarity, with folding angles of 7.3(2) and 8.1(2)° in TTF A and B respectively. Also, the presence of two TTF for one phenyl group allows now stronger S···S van der Waals interactions between TTF moieties with an intramolecular S···S distance of 3.56 Å, while the two TTF mean square planes make an angle of 64.9°. In the solid state, the molecules organize into layers (Fig. 4), with apparition of a criss-cross interaction between TTF moieties of neighbouring molecules and intermolecular S···S contacts as short as 3.87 Å.

Unit-cell view of (o-Me2TTF)2PPh (2).
The criss-cross overlap between TTF moieties in (o-Me2TTF)2PPh (2).
3.3 Structure of (o-Me2TTF)3P (3)
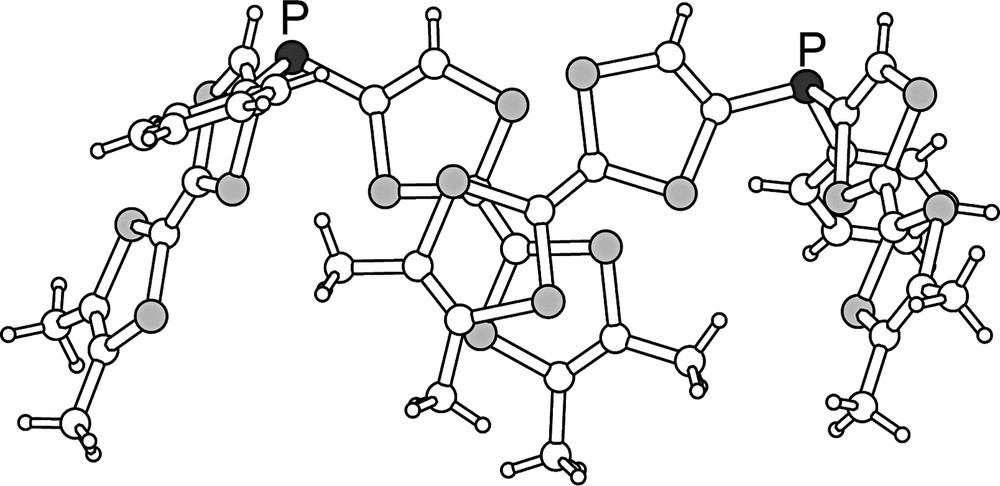
(o-Me2TTF)3P crystallizes in the trigonal system, space group R3c, with the molecule located on the three-fold axis (Fig. 5). Bond distances and angles compare with those described for o-Me2TTF as well as for (TTF)3P (Table 2). The two dithiole rings are now folded by 15.1(1) and 17.4(1)° for the dimethyldithiole and phosphorus-substituted dithiole rings, respectively. (o-Me2TTF)3P 3 organizes into columns along the c axis (Fig. 6), where both orientations of the propeller-shaped molecules alternate. This represents a rare example of transfer of the molecular symmetry to the crystal. Indeed, among the tertiary phosphines PR3 whose X-ray crystal structures have been determined and reported in the CSD, only six of them were found to crystallize on a three-fold symmetry axis (search has been conducted on the CSD November 2002 dataset, excluding structures with R1 value above 0.05; CSD codes corresponding to those phosphines are CANTEQ, PAMWAB, RICFEO, TAZPAD, TCYMPH and TPEPHP), and this number restrains to four [P(p-Tol)3 [9], P(9-anthryl)3 [10], P(CH2CN)3 [11] and P(C≡CPh)3] [12] if we remove those phosphines whose rigidity imposes a stringent three-fold molecular symmetry. Note also that R3c is one of the polar groups and that its occurrence is very rare in organic compounds. As a consequence, all phosphorus lone pairs are oriented along the same direction, giving to the crystal a net dipolar moment. The presence of polar axes has numerous implications on the chemical and physical properties of crystals [13], as the possibility for pyro-, piezo or ferroelectricity. Pyroelectricity was indeed reported recently in a rigid tertiary phosphine crystallizing in the polar R3m space group [14]. Electrocrystallization experiments, particularly with this phosphine and also with the corresponding phosphine oxide and sulphide are underway, in order to test their ability to keep these three-fold symmetry motifs in corresponding cation radical salts.

ORTEP view of (o-Me2TTF)3P (3). Thermal ellipsoids are drawn at 50% probability level.
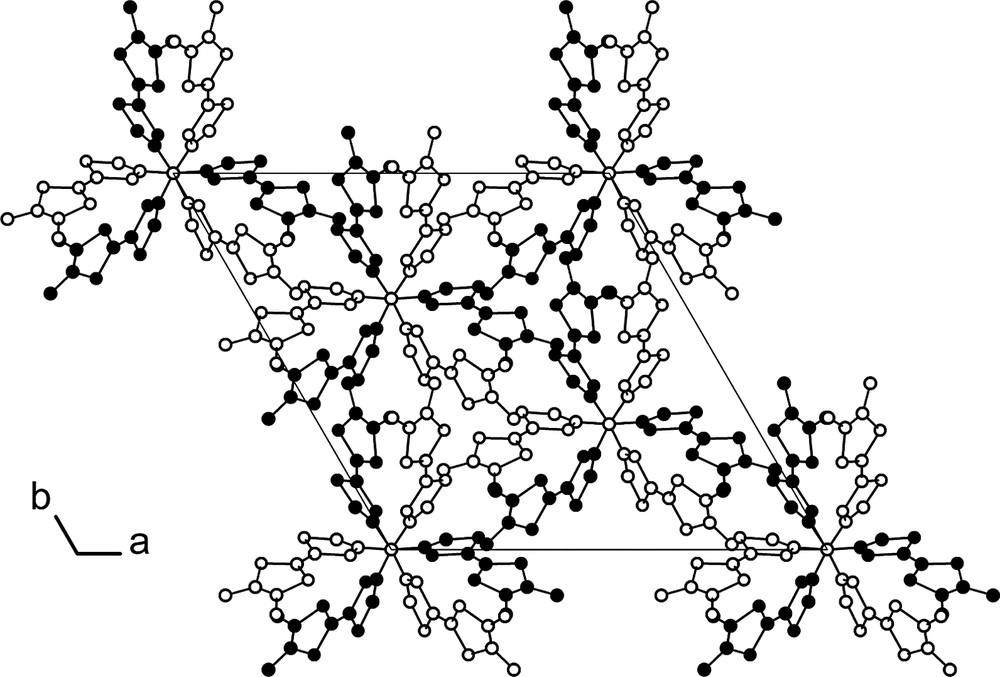
View of the unit cell of (o-Me2TTF)3P (3) along the c-axis. Both orientations of the propeller-like molecule are shown in white and black respectively. Phosphorus atoms are located on the three-fold axes.
Acknowledgements
Financial support from the French Ministry of Education and Research (to S.P.) is gratefully acknowledged.
Supplementary material
The supplementary materials concerning the X-ray crystal structures has been sent, as cif files, to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK, and can be obtained by contacting the CCDC and quoting the article details and depository numbers 231002–231004 for 1, 2 and 3, respectively.