

1 Introduction
As a result of recent advances in chemical biology and organometallic chemistry there is a need to develop efficient strategies for synthesis of P(III) phosphorus esters containing unusual additional ligands [1]. Esters bearing a fluorine ligand belong to this category. Phosphorus esters containing the P(III)–F moiety have been described in a variety of structural combinations but known methods are of limited value for the preparation of sensitive molecules derived from alcohols of biological importance and P chiral systems [2–6].
2 O-Arylphosphoramidites
Our interest in this type of structures has its roots in earlier studies on phosphitylating reagents holding two different leaving groups. Phosphoramidites fitted out with aryloxy ligands attached to the P(III) centre are of special interest [7–9]. In contrast to the amido group, which is activated under acidic conditions, the aryloxy group resistant to acid conditions can be exchanged by alcohols and other nucleophile under influence of strong bases without affecting the amido group. Phosphoramidites containing aryloxy leaving groups are exemplified by compounds 1, 2 and 3 (Fig. 1).
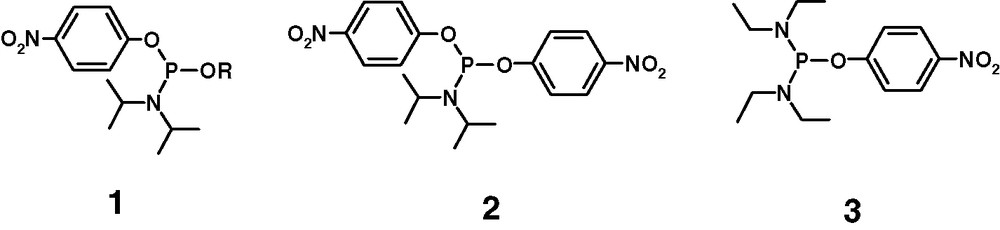
Representative phosphoramidites containing O-aryloxy group.
Compounds of these types are readily available from commercial starting materials and have been applied in the synthesis of natural and modified dinucleotides [7–9], sugars and izoprenoides [10]. Application of the phosphoramidite 1 (R = Me) in nucleotide chemistry and its dual reactivity is shown in Fig. 2.
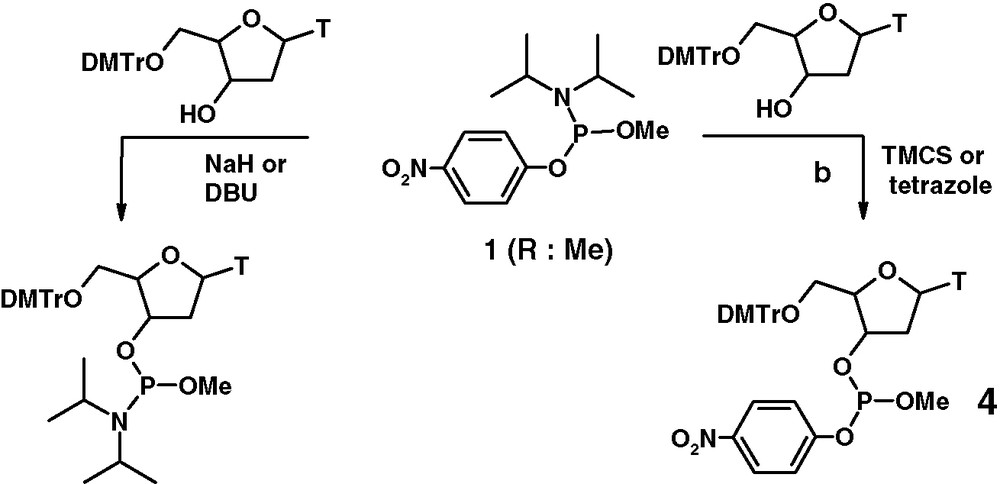
Dual behaviour of O-arylphosphoroamidite 1.
It has been demonstrated that the phosphitylating procedure described in Fig. 2 involves activation of P(III) isopropyloamino group under acidic conditions by tetrazole or trimethylchlorosilane (TMCS) (step b). In contrast, coupling of the phosphoramidite 1 by activation of the O-4-nitrophenoxy group (step a) requires intervention of strong bases such as DBU or sodium hydride. Phosphite 4 is formed in excellent yield and purity. Compounds of this type can be readily oxidized providing excellent access to the corresponding phosphates, thiophosphates, and selenophosphates.
A synthetic approach based on O,O′-bis-(4-nitrophenyl) N,N-diisopropylphosphor-amidite 2 (Fig. 3) has been already described in [11].
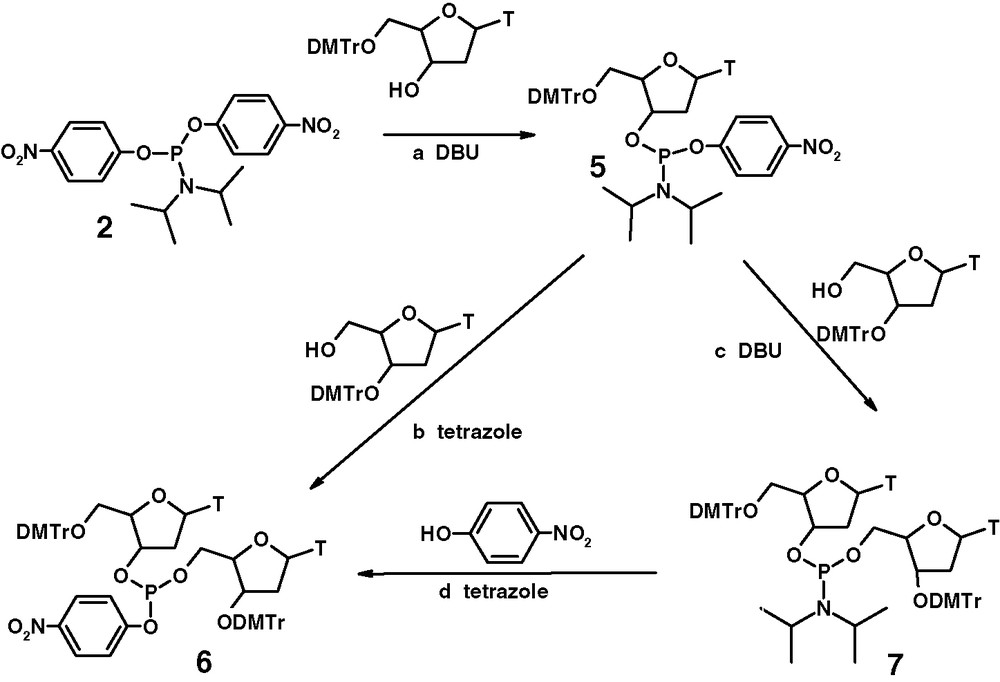
Synthesis of dinucleotide O-aryloxyphosphite.
The phosphitylation procedure starts by coupling of mononucleoside with the phosphitylating reagent in the presence of DBU (step a). The nucleoside O-4-nitrophenylphosphoroamidite 5 undergoes a second coupling via the N(i-Pr)2 group in the presence of tetrazole to give the dinucleoside arylphosphite 6 (step b). Phosphitylation described in step c leads to the dinucleoside phosphoroamidite 7, which is converted into the dinucleoside arylphosphite by reaction with 4-nitrophenol in the presence of tetrazole (step d). All these synthetic operations can be performed in a one-flask procedure in overall yield of the isolated product higher than 90%.
Dinucleoside O-arylophosphites can be also obtained starting from O-aryl phosphorodiamidites 3, as shown in Fig. 4 [12].
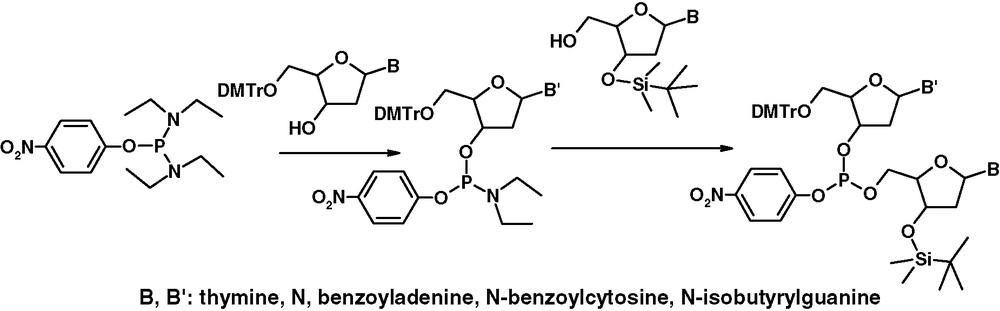
The alternative synthesis of dinucleoside O-aryloxyphosphite.
3 Synthesis of tricoordinate phosphorus containing P–F bond
We have devised a highly efficient strategy for the synthesis of P(III)–F systems based on replacement of a phenoxy group attached to a P(III) centre by a fluorine ligand [11–14] (Fig. 5).
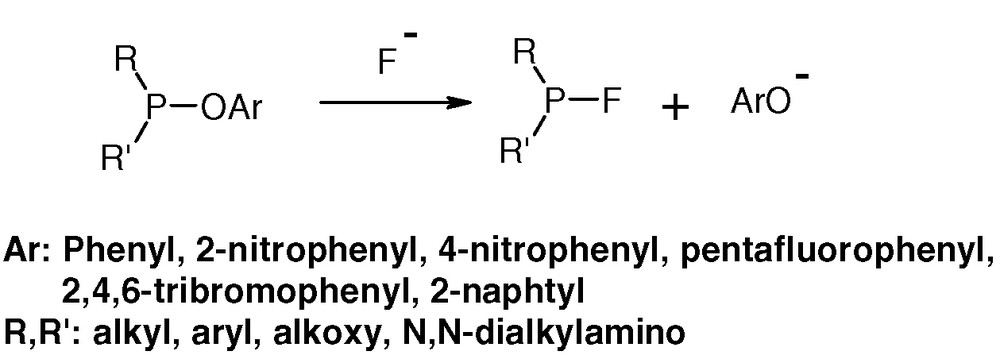
Conversion of P(III)–OAr compounds in to their P(III)–F analogues.
Appropriate source of fluoride anion are: KF or the KF-crown ether 18-system, CsF, nBu4NF and the best of all is tetrabutylammonium difluorotriphenylstannate. This transformation can be applied to a large variety of compounds, including those containing one of two ligands R, R' derived from alcohols of biological interest. The traditional synthetic route leading to P(III)–F compounds is via fluorination of the corresponding P(III)–Cl systems by reaction with SbF3, AsF3, NaF, CsF or potassium fluorosulfinate [2,3]. Other procedures have been used occasionally. Many of P(III)–F structures described before can be more conveniently prepared by our methodology.
Easy access to P(III)–OAr compounds allowed us to synthesize a number of P(III)–F structures derived from nucleosides and other alcohols of biological interest. The synthetic pathway leading to the dinucleosidylphosphorofluorides is shown in Fig. 6 [14].
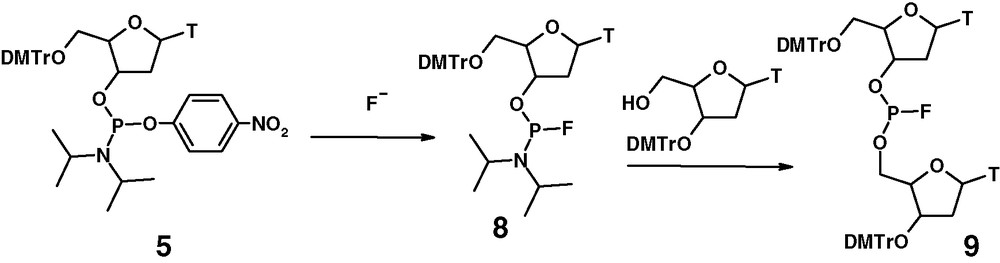
Synthesis of dinucleoside phosphorofluoridite.
Nucleoside bases are exemplified by thymine. The same sequence of reactions was applied for other nucleoside bases.
The nucleosidyl phosphoroamidofluoridites 8 are formed with some degree of stereoselectivity and can be separated into pure diastereoisomers. However, their coupling reaction with 3′-protected nucleosides in the presence of TMCS or tetrazole, under the usual conditions, affords dinucleosidyl phosphorofluoridite 9 as 1:1 mixtures of diastereomers in almost quantitative yields. It is noteworthy that this coupling does not affect the phosphorus–fluorine bond. Chromatography of phosphorofluoridites 9 on silica gel gives pure ‘fast’ isomers of high configurational stability. A typical 19 F NMR spectrum of the separated ‘fast’ diastereomers 9 is shown in Fig. 7. In the other class of P(III)–F compounds, 19F NMR spectroscopy is of great importance, often providing unambiguous information. The high purity of the ‘fast’ diastereoisomer 9 is also evident from 31P NMR spectroscopy.

19F NMR spectrum of the ‘fast’ diastereoisomer 9.
The configurational stability of phosphorofluoridites can be explained by the presence of the strong P–F bond and steric hindrance exerted by the nucleosidyl groups. Resolution of the racemic fluorophosphine MePhPF has been achieved only recently [15]. Trans 2-fluoro-4-methyl-1,2,3 dioxaphosphorinane has been prepared by Mikolajczyk et al. [16] Diastereoisomeric P(III)–F compounds 8 and 9 racemize in the presence of F– donors via achiral intermediates containing two fluorine ligands in apical positions [17]. Pure diastereoisomers 9 separated by chromatography were transformed in stereospecific way into the corresponding P(IV)–F compounds 10 or 11 by oxidation with selected oxaziridine [10,18] or bisbenzoyl disulfide [13] (Fig. 8). These transformations probably proceed with retention of configuration at the chiral phosphorus atom. Similar stable tetracoordinate diastereomeric dinucleotide phosphorofluoridates have been obtained earlier in our laboratory [19]. As expected, diastereomeric P(III)–F compounds 9, 10, and 11 are more resistant towards hydrolysis and other nucleophilic displacements than halogeno analogues. Hydrolitic susceptibility of compounds 9, 10, and 11 is strongly influenced by the presence of fluoride anions or silver cations. Similar behaviour was observed in respect of their stereochemical stability.

Stereoselective transformation of dinucleoside phosphorofluoridite into the corresponding fluorophosphate and thiofluorophosphate.
4 Phosphitylating reagents containing P(III)–F system
Our search for convenient synthesis of P–F structures derived from nucleosides and other alcohols of biological interest led to the preparation of novel efficient phosphitylating reagents in which the P–F bond is already present [20]. Two typical examples of reagents of this type are shown in Fig. 9.

Phosphorofluoroamidites 12 and 13.
Namely [O-(tert-butyl) N,N-diisopropylfluorophosphoramidite F-P(NiPr2)OtBu] 12 and [O-(2-cyanoethyl) N,N-diisopropylfluorophosphoramidite F–P(NiPr2)OCH2CH2CN] 13. They can be prepared from commercial starting materials: N, N-diisopropylaminodichlorophosphine or O-(2-cyanoethyl) N,N-diisopropylchlorophosphoramidite in very high yield and purity (Fig. 10). The relative stability of these compounds towards hydrolysis and oxidation is advantageous in synthesis procedures.

Preparation of phosphorofluoroamidites 12 and 13.
Phosphitylating reagents 12 and 13 react with alcohols in the presence of activators such as tetrazole [21,22], trimethylchlorosilane (TMCS) [14] or 2,4-dinitrophenol [23] (Fig. 11). The phosphorus–fluorine bond is not affected. TMCS proves to be a better activator than tetrazole, which must be used in large excess. The strongly electronegative fluorine does not affect the use of the activated aminoligand as a leaving group in phosphitylation procedures. This behaviour differs from that of P(III) amidites containing the highly electronegative CF3 group attached to the phosphorus centre. Compounds of this type are immune to activation by tetrazole or TMCS [24].
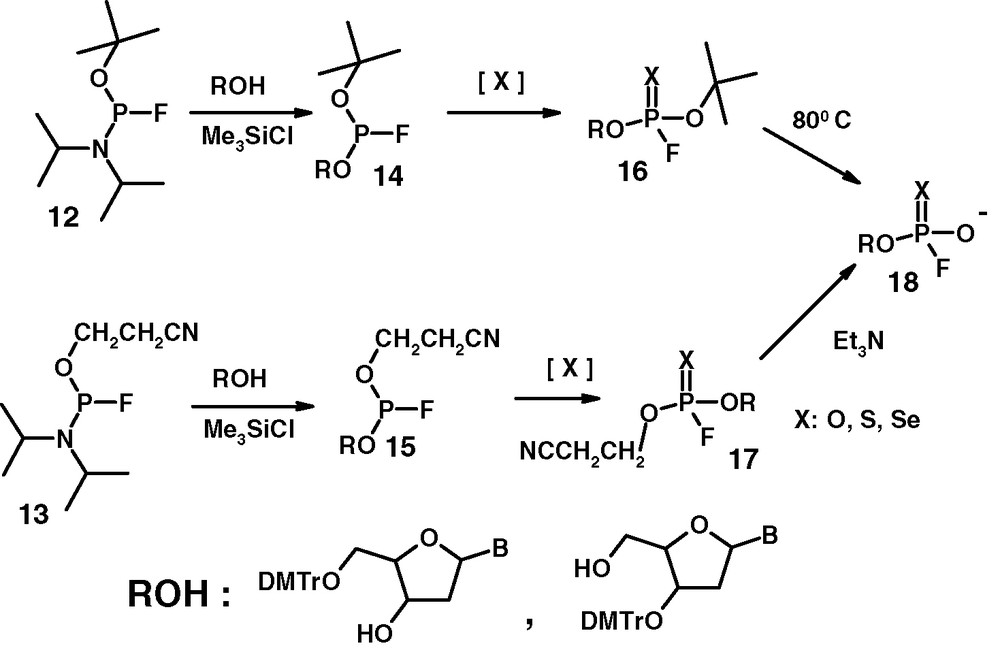
Synthesis of fluorophosphates 18 via reagents 12 and 13.
Nucleosidyl phosphorofluoridates 18 were originally obtained by Wittmann [25] and used in biological studies [26]. The thioanalogues of 18 (X = S) have been prepared for the first time in this laboratory. Our phosphorylation procedure leading to Wittmann's type of compounds is illustrated by the reactions of 5′-O-DMTr and 3′-O-DMTr protected thymidines with phosphoroamidites 12 and 13 followed by oxidation, sulphurization or selenization of the intermediate phosphorofluoridites 14 and 15. This transformation is performed by tert-butyl hydroperoxide, elemental sulphur or elemental selenium. In the final step the tert-butyl group undergoes thermal elimination and the 2-cyanoethyl group is removed by β-elimination under standard conditions to give the desired phosphorofluoridates or their structural thio and seleno analogues. The sequence of reactions leading to compounds 18 (X: O, S, Se) is shown in Fig. 11. The DMTr protective group at the 5′-O or 3′-O centre is not removed under these conditions.
5 TMCS as an alternative activating reagent
In our studies on the synthesis of P(III)–F nucleotides, we were confronted by the challenge of finding a mode of activation using compounds other than tetrazole or similar. This requirement was connected with low efficiency of classic activators. Our earlier work on interaction of P(III) amides with halogenosilanes suggested that phosphoramidites would react with alcohols in the presence of trimethylchlorosilane (TMCS) as a catalyst [27]. The reaction of P(III) amidites with an equivalent amount of nucleoside proceeds in the presence of TMCS in very high yield and at rates comparable with or higher than those when tetrazole is used. Phosphitylations activated by TMCS proceed at room temperature in solvents like THF, CH2Cl2 or MeCN. On average, the amount of activator required for an efficient coupling is ca 30–60% of the stoichiometric ratio. It is most likely that this activator acts via formation of a salt-like intermediate in its reaction with the phosphoramidite.
6 Synthetic application of P(III)–F compounds
As anticipated, P(III)–F compounds are less prone toward nucleophilic displacement than their I, Br, Cl analogues. However they can serve as valuable, easy-to-handle substrates, in special synthetic procedures.
For example 3′-thymidine-O-methyl-N,N-diisopropylphosphoroamidite has been prepared by condensation of lithium methoxide with the corresponding fluorophosphoro-amidite [10] (Fig. 12).
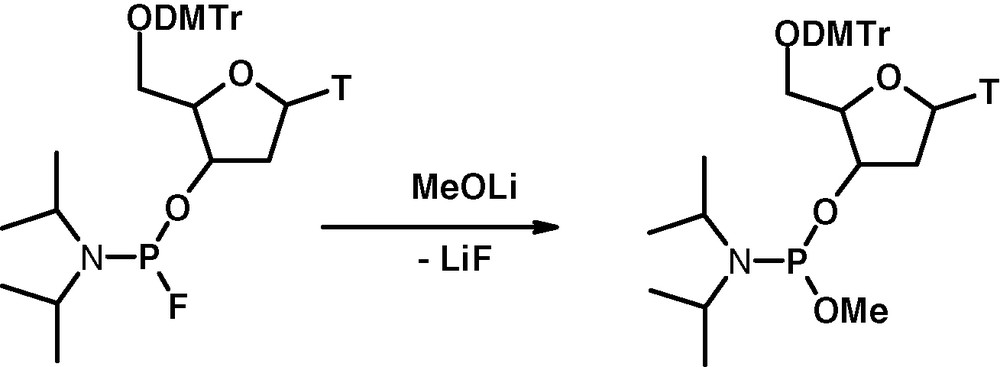
Synthesis of O-Me phosphoroamidite.
As shown in several examples, trimethylchlorosilane (TMCS) acts as a highly efficient catalytic activator for the replacement of a PIII–NR2 amino group by alcohols to form the corresponding esters. We have noticed that PIII–F compounds can also react as reagents for the replacement of a fluorine ligand by an appropriate alcohol when at least 1 equiv of TMCS is used (Fig. 13). This reaction is relatively slow. Its driving force is the formation of trimethylfluorosilane [28].
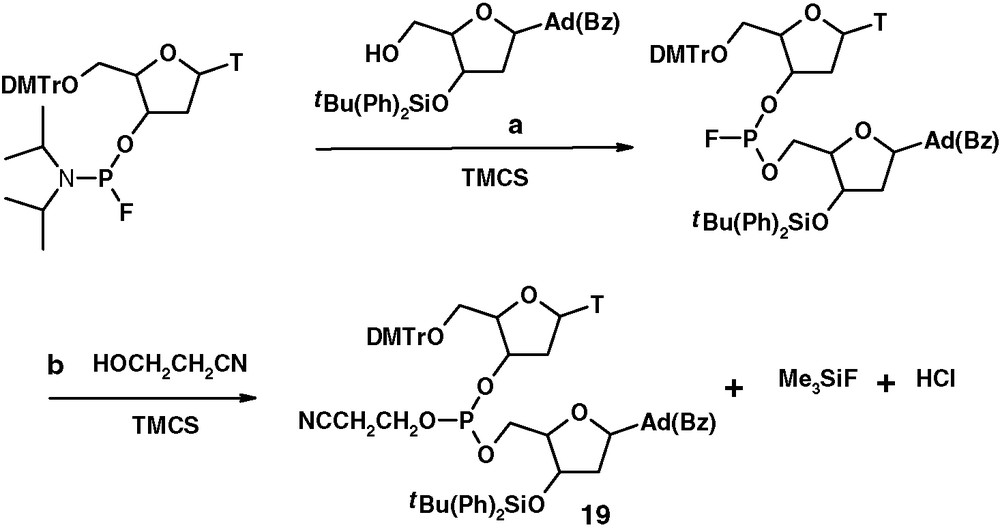
Alternative synthesis of tricoordinated phosphate via replacement of fluoro group.
In the sequence of reactions shown above, TMCS acts in step a as catalytic activator in the coupling leading to the dinucleoside phosphorofluoridite. In step b, the same compound acts as a reagent to give the corresponding phosphite 19, Me3SiF and HCl. Hydrogen chloride formed can be trapped by 4-Å molecular sieves or trialkylamine.
Replacement of the fluorine ligand by an alkyl (aryl) group in the action with organometallic compounds is of special interest. Promising results have been obtained with Grignard reagents in the synthesis of new types of P(III) ligands in a stereoselective way and with high chemical yields [10] (Fig. 14).
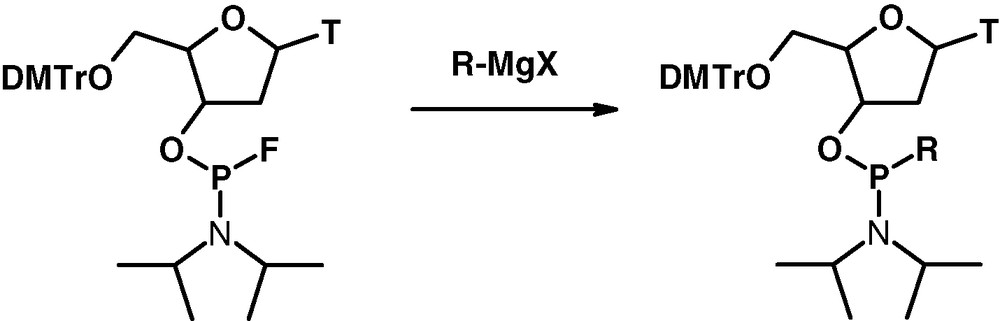
Synthesis of nucleoside phosphonoamidites.
Easy access to P(III)–F compounds allowed us to work out the new and convenient synthesis of P(III)–CF3-type species in a variety of structural combinations [28]. We have discovered that the P(III)–F reacts with the (trifluoromethyl)trimethylsilane (Me3SiCF3, Ruppert reagent) [29]. This reaction, catalysed by fluoride anions, proceeds at room temperature in THF or acetonitrile in very high yields [30]. Caesium fluoride is a convenient fluoride anion donor, but other donors like TBAF can also be used.
A highly useful observation has been made that the formation of P(III)–F compounds from the corresponding P(III)–aryloxy starting materials can be combined with the reaction of P(III)–F species leading to P(III)–CF3 structures (Fig. 15).
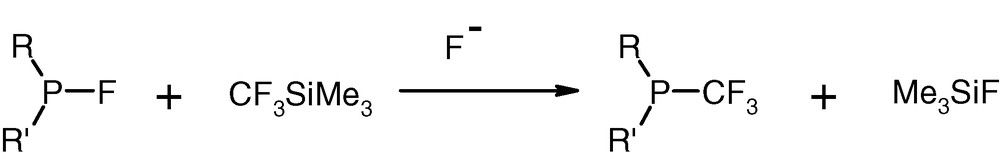
Synthesis of P(III)–CF3 compounds.
This reaction can be a part of sequential procedure that combines formation of P(III)–F compounds with transformation into the corresponding P(III)–CF3. An example of the sequential procedure is the synthesis of bis (trifluoromethyl)(diisopropylamino)phosphine.
This mode of proceeding has been applied to a variety of P(III)–F structures containing alkoxy, alkylthioxy, amino and aryl ligands. The corresponding P(III)–CF3 structures are obtained in excellent yield including modified nucleotides holding P(III)–CF3 moiety (Fig. 16).
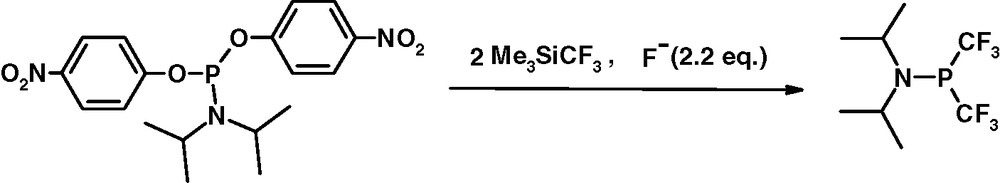
Synthesis of bis (trifluoromethyl) (diisopropylamino)phosphine
For the reaction shown in Fig. 15, the catalytic cycle (Fig. 17) is proposed.
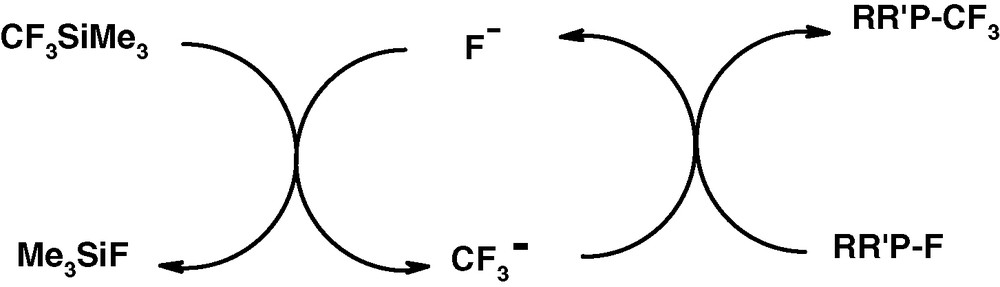
Catalytic cycle leading to P(III)–CF3 structure.
In this cycle the strength of the P–F bond is compensated by the high affinity of fluorine for the silicon centre.
Acknowledgments
The authors' work was supported by the State Committee of Scientific Research of Poland (Grant No. 7 T09A 155 21) and the Polish-German project (grant POL 01/014).