

1 Introduction
Les inhibiteurs de la transcriptase inverse et de la protéase constituent la thérapie antivirale de choix dans le traitement de l’infection par le virus de l’immunodéficience humaine [1]. Ces deux cibles jouent un rôle crucial dans le cycle de réplication de VIH-1. La combinaison d’un inhibiteur de la protéase avec deux inhibiteurs de la transcriptase inverse (trithérapie) a permis de retarder la progression de l’infection vers le stade de syndrome de l’immunodéficience acquise (SIDA) et d’améliorer la qualité de vie des patients [2]. Par ailleurs, les effets secondaires [3,4] (anémie, perte de poids, les infections opportunistes dues à l’utilisation prolongée) ainsi que le phénomène de résistance [5,6] (dues au taux de mutation particulièrement élevé chez le virus) font que la recherche de nouveaux inhibiteurs reste une solution à envisager [7–9]. Les inhibiteurs de la protéase actuellement utilisés présentent plusieurs inconvénients (faible biodisponibilité, due au grand poids moléculaire, problème pharmacocinétique principalement du à la nature des liens peptidiques et à la présence de plusieurs centres stéréogéniques) [10].
Dans notre laboratoire, la stratégie adoptée pour l’élaboration d’inhibiteurs non peptidiques est basée sur l’obtention d’un composé de faible masse molaire. En général, les produits de masse molaire inférieure à 500 g mol–1 ont de grandes capacités à traverser efficacement les membranes cellulaires. Associée au caractère non peptidique, cette caractéristique favorise la biodisponibilité du composé. Les travaux réalisés antérieurement dans notre laboratoire [11] ont conduit à la synthèse de composés possédant une fonction énol difonctionnalisée (Fig. 1) ayant un groupement hydroxyle très polarisé, qui favorise les interactions avec l’enzyme.
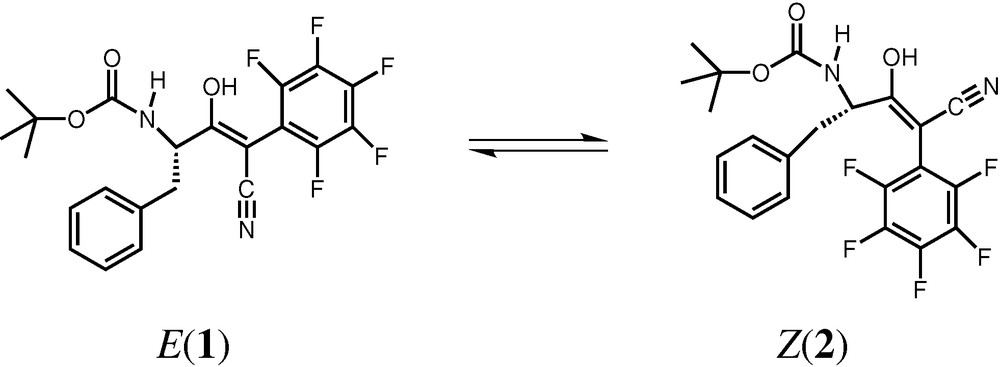
Énols acycliques.
Des études de structure–activité sur ces énols difonctionnalisés [12] ont montré que le groupement protecteur de l’amine, terbutoxy carbonyl (Boc), le groupement benzyle (chaîne latérale de Phe ou de Tyr) ainsi que les substituants pentafluorophényle et nitrile représentent la meilleure combinaison de cette famille de molécules pouvant inhiber la protéase du VIH-1 (Ki = 10 μM) en optimisant l’affinité envers la protéase. Ces composés (énols difonctionnalisés) peuvent exister sous deux formes énolique et cétonique. Des études de résonance magnétique nucléaire (RMN) sur ces composés ont montré que ces derniers existent essentiellement sous la forme énol dans un mélange de deux isomères géométriques E(1) et Z(2) en équilibre (Fig. 1). La modélisation moléculaire sur le meilleur inhibiteur énol de cette série a montré que l’énergie d’interaction du complexe protéase-E-énol est de 12 kcal mol–1 (inférieure à celle du complexe protéase-Z-énol). Le but de notre travail est d’évaluer l’impact sur l’affinité pour la protéase du VIH-1 de la combinaison de ces fonctions (énol avec deux petits groupements électro-attracteurs hydrophobes donnant un hydroxyle polarisé) dans un système rigide. La formation d’une structure cyclique à six chaînons de type énol-lactone permet d’orienter spécifiquement dans l’espace les groupements de l’énol qui permettent l’obtention, entre autres, d’un composé très similaire à l’énol acyclique maintenu sous sa forme E et susceptible de présenter un potentiel d’inhibition plus important. Dans un premier temps, le composé énol-lactone cyclique constituera la molécule cible à préparer. Afin de réaliser une étude de relation structure/activité et de déterminer la meilleure combinaison de substituants pour l’inhibition de la protéase du VIH, d’autres énol-lactones comportant différents substituants seront préparées. Enfin, une synthèse énantiosélective d’énol-lactone a été réalisée.
2 Résultats et discussions
2.1 Synthèse des énol-lactones α,δ-substituées
L’approche utilisée consiste à préparer l’énol-lactone peu substituée suivie de l’introduction des substituants requis sur la lactone. La construction du cycle lactonique a été réalisée aisément en deux étapes selon la méthode de Huckin [13].
Le cycle étant formé, nous avons introduit différents substituants en position α de l’énol-lactone 3 en commençant par le groupement pentafluorophényle, qui donne, en effet, les meilleurs résultats d’inhibition sur l’énol acyclique [11]. Nous nous sommes inspirés des conditions de réaction déjà rapportées dans la littérature permettant d’effectuer des α-alkylations de l’hydroxycyclohexénone pour donner les produits α-substitués correspondants 4 [14].
Suivant ces procédures, la lactone 3 est traitée par l’hydroxyde de benzyltriméthylammonium et le bromure de pentafluorobenzène (Fig. 2). Nous avons constaté que le seul produit que nous avons isolé du milieu réactionnel était l’α-benzyl énol-lactone 5, avec un rendement de 7%, plutôt que l’α-pentafluorophényl énol-lactone attendue. Il est connu que le réactif utilisé, l’hydroxyde de benzyltriméthylammonium, comprend une certaine quantité de benzyl méthyl éther (BnOMe), qui est susceptible de réagir avec l’énol-lactone énolisée pour donner le benzyl énol-lactone 5. Le professeur Zdenek Valenta de l’université du Nouveau-Brunswick (communication personnelle) a déjà observé ce type de réaction secondaire sur des cyclohexadiones. Des essais visant à augmenter le rendement de la réaction en variant la température ou la quantité d’eau (modification de la proportion d’eau/méthanol) n’ont pas donné le résultat escompté.
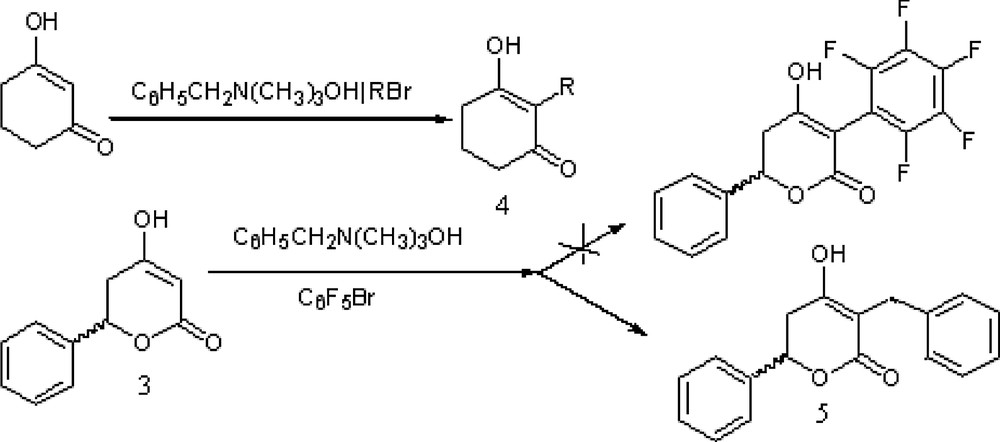
Alkylation de l’énol-lactone 3.
Les essais cinétiques enzymatiques avec l’énol-lactone 5 ont donné une constante d’inhibition très encourageante de 1,6 μM. Nous nous sommes donc intéressés à introduire le groupement benzyle (groupement hydrophobe) en optimisant dans un premier temps les rendements de la réaction de benzylation. Dans un second temps, nous avons envisagé de préparer des analogues renfermant différents groupes aryles. Ainsi, le traitement de la lactone 3 avec le bromure de benzyle en présence du tert-butoxyde de potassium dans le diméthylsulfoxyde [15] donnèrent l’α-benzyl énol-lactone 5 désiré, avec un rendement acceptable de 62%. La structure du composé 5 est confirmée par le spectre RMN 1H, qui met en évidence la présence des signaux correspondant aux protons du groupe benzyle (un singulet à 3,55 ppm intégrant pour deux protons, attribuable au groupe méthylène). Notons que deux autres produits ont été isolés et identifiés comme produits de la O-alkylation 6 et de la O,C-alkylation 7. Nous avons fait des études sur la formation de ces produits secondaires [16]. Pour étudier l’influence de la nature des substituants portés par le groupe benzyle sur les interactions de l’énol-lactone avec le site actif et ainsi augmenter le potentiel d’inhibition, quelques énol-lactones α-benzyl ont été préparées. Les conditions de Balog [15] ont été utilisées pour introduire des benzyles comportant différents substituants par l’intermédiaire des halogénures benzyliques correspondants. Les structures des énol-lactones 5 et 8–11 ainsi que leur rendement sont indiquées dans le Tableau 1. Ces énol-lactones α-substituées existent essentiellement sous la forme énol dans le chloroforme deutérié ou le diméthylsulfoxyde. En effet, leurs spectres de RMN 1H présente en particulier un signal large à environ 10 ppm, attribuable au groupement hydroxyle de la fonction énol, et l’absence du signal du proton méthine, qui serait normalement attendu pour le tautomère cétonique.
Rendements et constantes d’inhibition des aryl énol-lactones 7–11
| Numéro | Ar | Rendement | Kia (μM) (sol.b μM) |
| 5 | 62% | 1,2 (100) | |
| 8 | 49% | 4,9 (75) | |
| 9 | 45% | 140 (50) | |
| 10 | 53% | 3,2 (100) | |
| 11 | 57% | 12 (25) |
a Constante d’inhibition avec une marge d’erreur de 0,1 μM.
b Sol. : solubilité.
Ainsi, l’α-bromoxylène conduit à l’énol-lactone 8 avec un rendement de 49%, et le spectre RMN du proton présente, outre les signaux associés aux protons benzyliques, un singulet à 2,49 ppm correspondant aux trois protons du groupe méthyle. Le groupement p-trifluorométhylbenzyle est introduit avec 45% de rendement (Fig. 3).
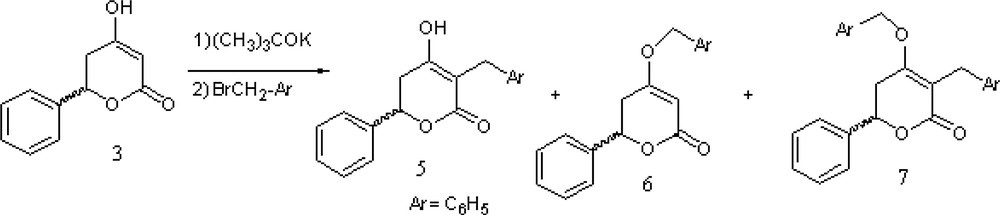
Introduction de groupement benzyle avec divers substituants.
2.2 Résultats des tests enzymatiques et discussion en terme de relation structure–activité
2.2.1 Méthodologies des essais enzymatiques
L’évaluation des produits de synthèse comme inhibiteurs potentiels de la protéase du VIH-1 a été faite à partir de méthodes enzymatiques connues de la littérature [17]. Nous avons utilisé la méthode de fluorescence faisant appel à un substrat fluorogénique développé par Matayoshi et al. [18] en l’adaptant à notre instrumentation disponible au laboratoire, i.e. un spectrophotomètre UV/visible Caryl transformé en fluorimètre par l’ajout d’un accessoire à l’appareil. Ce test est très rapide, de sorte que trois à six inhibiteurs peuvent être évalués chaque jour de façon précise et fiable. Le principe de la méthode repose sur l’utilisation d’un substrat modifié de la protéase du VIH comprenant deux groupements particuliers de part et d’autre du lien peptidique hydrolysé.
Nous distinguons, dans le substrat H–Arg–Glu(EDANS)–Ser–Gln–Asn–Tyr–Pro–Ile–Val–Gln–Lys(DABCYL)–Arg–OH utilisé dans nos tests et vendu par la compagnie Molecular Probes, un groupement EDANS, ainsi qu’un groupement DABCYL, qui, lorsqu’ils sont liés au même peptide, participent à un système de transfert d’énergie en résonance « RET ». De cette façon, lorsque la solution est soumise à un faisceau lumineux, la lumière fluorescente émise par l’EDANS est absorbée par le groupement DABCYL.
Lorsque le peptide est scindé par l’action de la protéase, le système d’absorption intramoléculaire devient inopérant, rendant la solution fluorescente. De cette façon, meilleur est l’inhibiteur, moins forte sera l’augmentation de la fluorescence émise pour une concentration équivalente. Par des calculs appropriés [19], il est possible de déterminer la constante d’inhibition (Ki) d’un produit. Plus cette valeur est faible, plus l’inhibiteur est efficace, ce qui se traduit par une moins grande quantité de produit nécessaire pour inhiber la protéase. Les excellents inhibiteurs ont des Ki de l’ordre du nM. Toute molécule, faisant partie d’une nouvelle famille pouvant inhiber la protéase avec des Ki de l’ordre du μM a habituellement un intérêt et considérée chef de file (lead compound). D’autres modifications peuvent alors être apportées à la molécule en question pour obtenir des inhibiteurs améliorés.
2.2.2 Potentiel de l’énol-lactone comme inhibiteur de la protéase du VIH
Les essais de cinétique enzymatique avec l’énol-lactone 5 ont donné une constante d’inhibition de 1,6 μM. L’amélioration de l’activité est évaluée à 6 fois par rapport au produit acyclique 1 ou 2. Notons que sans le groupe benzyle l’activité serait de l’ordre de 178 μM. Ceci pourrait s’expliquer par la forme du site actif, qui contient plusieurs sous-sites où les substituants doivent se loger pour interagir avec la protéase. Ce résultat prometteur nous a incités à poursuivre l’étude de cette nouvelle famille de composés. L’ajout de différents substituants sur le benzyle tend à augmenter l’inhibition de ces dérivés envers la protéase du VIH-1. Les dérivés préparés 8–11 sont rapportés avec leurs constantes d’inhibition respectives dans le Tableau 1. Nous avons escompté que certains substituants sur le benzyle pourraient favoriser une meilleure interaction avec la protéase du VIH-1. Ce n’est pas le cas avec des substituants tels que p-méthyle 8 (Ki = 4,9 μM), p-trifluorométhyle 9 (Ki = 140 μM), p-fluoro 10 (Ki = 3,2 μM) et pentafluoro 11 (Ki = 12 μM).
En effet, le remplacement d’un hydrogène en position para sur le cycle benzylique par un groupement méthyle, un groupement trifluorométhyle ou un atome de fluor fait chuter l’activité inhibitrice d’un facteur de 3, de 9 et de 2 respectivement. Les quelques exemples indiqués dans le Tableau 1 suggèrent que, dans cette série d’inhibiteurs, l’ajout de substituants sur le benzyle est plutôt défavorable à l’inhibition, contrairement à d’autres inhibiteurs rapportés dans la littérature, où on observe une augmentation de l’inhibition. Par exemple, dans certains cas, l’atome de fluor peut établir un pont hydrogène avec des acides aminés polaires à proximité dans la cavité de l’enzyme.
3 Synthèse énantiosélective
Durant la réalisation de nos travaux, Romines et al. [20] publièrent des résultats sur l’hydroxypyrone 12, dont la structure est analogue à celle de notre énol-lactone 5. Les principales interactions obtenues d’une structure cristalline du complexe formé entre l’hydroxypyrone 12 et la protéase sont présentées sur la Fig. 4.

Comparaison de la structure de notre énol-lactone 1 avec celles connues de la pyrone.
On observe que la fonction pyrone joue un rôle important en remplaçant la molécule d’eau présente normalement dans le site actif de la protéase. Le noyau pyrone permet l’établissement des ponts hydrogènes entre l’inhibiteur et les ILe50 de la partie mobile de la protéase. Vu la similitude des structures 5 et 12, nous pouvons nous attendre à une complexation semblable de la part de nos énol-lactones au site actif de la protéase.
3.1 Résultats et discussions
Des études plus approfondies des structures cristallines des molécules les plus prometteuses ont permis de montrer que le groupement hydroxyle dans les systèmes hydroxy-pyrone interagit avec les acides aspartiques 25 et 25′ de la protéase, alors que la fonction pyrone interagit avec les isoleucines 50 et 50′. C’est la disposition de part et d’autre du cycle ainsi que la nature des substituants qui permettent d’établir de fortes interactions avec l’enzyme. Contrairement à la plupart des inhibiteurs acycliques connus de la protéase du VIH-1, les pyrones ne nécessitent pas la présence d’une molécule d’eau dans le site actif. Les mêmes types d’interactions sont attendus pour la série des énol-lactones. Dans les travaux mentionnés [20], le type des molécules proposées comporte un énol conjugué avec une fonction lactone, ce qui confère à l’hydroxyle une forte polarisation et devrait favoriser les interactions avec les acides aspartiques 25 et 25′ du site actif de la protéase. L’hydroxyle de notre énol-lactone est beaucoup plus polarisé que celui des hydroxy pyrones et des hydroxy coumarines rapportées, étant donné que ces dernières sont liées à des cycles aromatiques dont l’effet polarisant est moins important.
Nous avons minimisé les structures de l’α-benzyl δ-phényl énol-lactone 5, des énantiomères R et S ainsi que celle de la pyrone 12. Les trois structures représentées sur la Fig. 2 ont été minimisées grâce aux logiciels Insight et Discover de Biosym Technologies Inc. et à la station graphique Elan R4000 de Silicon Graphies (Figs. 5 et 6).
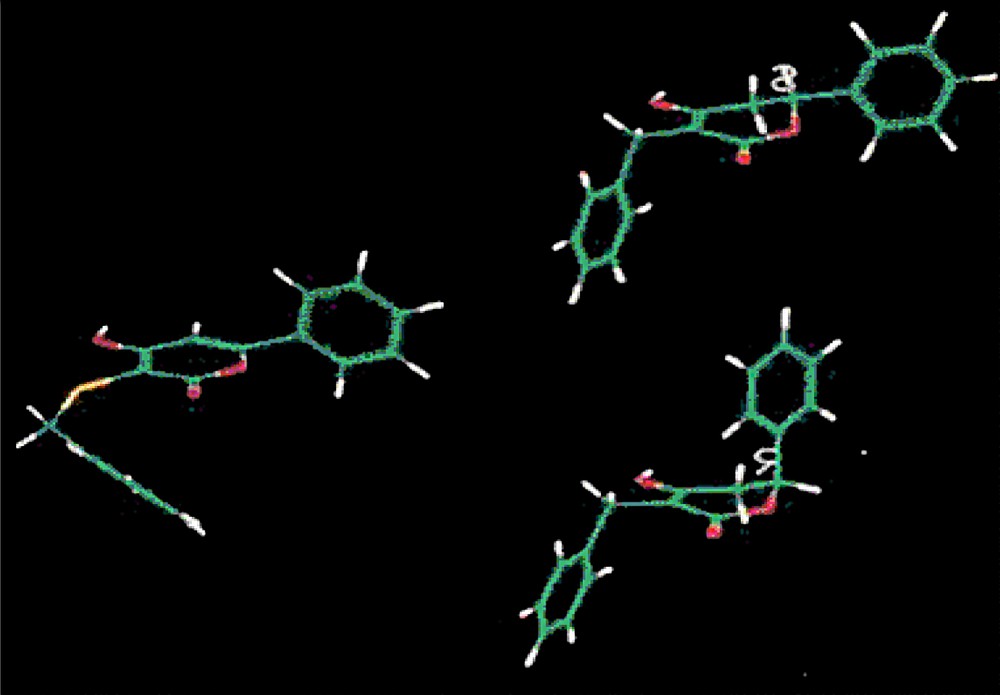
Structures minimisées de la pyrone 12 (à gauche) et des deux énantiomères R (à droite en bas) et S (à droite en haut) de l’énol-lactone 5.
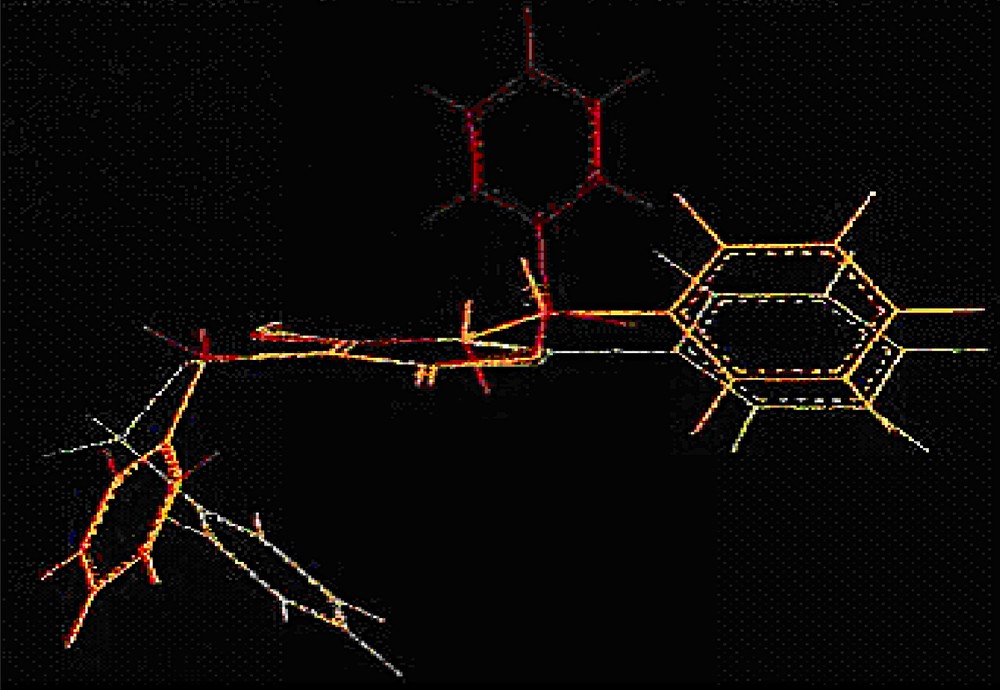
Structures minimisées et superposées de la pyrone 12 (en blanc) et des deux énantiomères S (en jaune) et R (en rouge) de l’énol-lactone 5.
À première vue, la conformation adoptée par l’énantiomère S de l’énol-lactone est plus semblable à la pyrone (à gauche) que celle de l’isomère R. Ceci est plus clair dans la Fig. 3, où nous avons superposé les trois structures. Nous remarquons qu’avec l’énantiomère R (en rouge), après superposition des cycles lactone et pyrone, que le groupement phényle de énol-lactone n’a pas la même orientation dans l’espace que celui de la pyrone. En revanche, l’énantiomère S (en jaune) possède une bonne corrélation comme le démontre le haut degré de superposition à la pyrone (en blanc). En principe, un des deux énantiomères de l’énol-lactone, en particulier, l’isomère de configuration S devrait être potentiellement plus actif.
Étant donné que nous avons un mélange des deux énantiomères R et S, une synthèse en vue de l’obtention d’énol-lactones optiquement pures s’impose. Plusieurs approches ont été tentées et la suivante est celle qui a donné deux énantiomères avec le meilleur excès émantiomérique. Les énol-lactones 13 et 14 sont obtenus selon la méthode de Carreira et al. [21]. L’alkylation en α des β-cétolactones 15 et 16 est réalisée avec le bromure de benzyle en présence du tert-butoxyde de potassium dans le diméthylsulfoxyde [15]. L’énantiomère S 15 (Ki = 1,5 μmol) semble démontrer une meilleure affinité avec le site actif que l’énantiomère R 16 (Ki = 1,8 μmol), ce qui confirme notre hypothèse de départ.

4 Conclusion
À la suite des travaux de Vaillancourt dans notre laboratoire, nous avons émis l’hypothèse que l’inhibition de la protéase par des énols acycliques pouvait être augmentée lorsque ces derniers seraient incorporés dans un cycle (par exemple, énol-lactones) représentant la fonction énol acyclique avec la configuration E, vraisemblablement plus active que l’isomère de configuration Z. L’ensemble de ces caractéristiques devait permettre une meilleure interaction entre l’inhibiteur et le site actif de la protéase. Les composés de type énol-lactone ont été obtenus. L’évaluation du potentiel d’inhibition de ces nouveaux composés envers la protéase a été effectuée.
La δ-phényl-α-benzyl énol-lactone 5 avec une constante d’inhibition de 1,6 μM s’avère une molécule inhibitrice prometteuse. L’activité de cette énol-lactone 5 est six fois supérieure à celle de l’énol acyclique 1 et 2. La fonction énol-lactone apparaît comme élément structural important pour l’activité. Ceci est dû probablement à la conjugaison de l’énol avec la lactone, ce qui permet de polariser efficacement l’hydroxyle de l’énol.
La séparation des deux énantiomères R et S de l’énol-lactone a été réalisée. L’isomère de configuration S (phényle) 15 semble établir des interactions favorables avec le site actif de la protéase, conformément avec l’étude comparative de modélisation moléculaire effectuée sur nos énol-lactones et la pyrone de la littérature.
5 Partie expérimentale
5.1 Notes générales
Les spectres de résonance magnétique nucléaire du proton (1H RMN) et du carbone (13C RMN) ont été enregistrés sur les appareils Bruker ANIX de 400 MHz (université de Montréal) et de 500 MHz (institut Armand-Frappier), et Varian Gemini 300 MHz (université du Québec à Montréal). Le deutériochloroforme (CDC13) est le solvant normalement utilisé pour les échantillons de RMN, sauf dans les cas où les produits ne sont pas solubles. Le diméthylsulfoxyde-d6 est alors utilisé comme solvant deutérié. Dans tous les cas, le tétraméthylsilane (TMS) a servi de référence interne (δ = 0) pour établir les valeurs de déplacement chimique δ exprimées en parties par million (ppm). Les points de fusion ont été déterminés dans des tubes capillaires sur un appareil Büchi 530 et ne sont pas corrigés. Quant aux spectres infrarouges (IR), ils ont été enregistrés dans le chloroforme entre deux pastilles de chlorure de sodium sur un spectrophotomètre infrarouge–FT Bomen Michelson 100.
5.2 Procédure générale
5.2.1 (6 RS)-5,6-Dihydro-4-hydroxy-3-benzyl-6-phényl-2H-pyran-2-one 5
À une solution de la lactone 3 (50 mg, 0,26 mmol) dans le diméthylsulfoxyde (2 ml) est ajouté le tert-butoxyde de potassium (32 mg, 0,29 mmol), puis le bromure de benzyle (32 μl, 0,26 mmol). Le mélange réactionnel est agité pendant une nuit à température ambiante puis la saumure (10 ml) est ajoutée. Le mélange est extrait avec l’acétate d’éthyle (2 × 20 ml). La phase organique est séchée sur MgSO4 puis concentrée sous pression réduite. Le brut est chromatographié pour donner le produit d’alkylation 5 (76 mg, 62%) sous la forme d’un solide blanc. Rf : 0,3 (AcOEt/hexane: 50:50). IR, (KBr): 3036, 2896, 1796, 1590, 1498, 1292, 1008 cm–1. pf. 176 °C. RMN 1H (300 MHz, DMSO-d6) δ 2,71 (dd, J1 = 17,1 Hz et J2 = 4,0 Hz, 1H, CHH–C–OH), 2,97 (dd, J1 = 17,1 Hz et J2 11,9 Hz, 1H, CHH–C–OH), 3,48 (s, 2H, CH2–Ar), 5,45 (dd, J1 = 11,9 Hz et J2 4,0 Hz, 1H, CH–Ph), 7,09–7,50 (m, 10 H, H Ar), 11,03 (s, 1H, OH). RMN 13C (75 MHz, DMSO-d6) δ 28,56, 34,62, 75,26, 101,78, 125,44, 126,36, 127,96, 128,14, 128,24, 128,42, 139,17, 140,96, 166,57, 167,57.
5.3 Synthèse de dérivés de la lactone
5.3.1 (6 RS)-5,6-Dihydro-4-hydroxy-3-(4'-méthylbenzyl)-6-phényl-2H-pyran-2-one 8
Suivant la procédure générale, sauf en utilisant le α-bromo-p-xylène (51 mg, 0,28 mmol), le produit d’alkylation 8 est obtenu (38 mg, 49%) sous la forme d’un solide blanc. Rf: 0,1 (AcOEt/hexane: 50:50). IR, (KBr): 3068, 1744, 1628, 1369, 1088, 837, 696 cm–1. pf. 115 °C. RMN 1H(400 MHz, DMSO-d6) δ 2,49(s, 3H, CH3), 2,73 (dd, J1 = 18,0 Hz et J2 = 3,1 Hz, 1H, CHH–C–OH), 2,99 (dd, J1 = 18,0 Hz et J2 = 12,4 Hz, 1H, CHH–C–OH), 3,63 (s, 2H, CH2–Ar), 5,48 (dd, J1 = 12,4 Hz et J2 = 3,1 Hz, 1H, CH–O), 7,34–7,60 (m, 9H, H–Ar), 11,25 (s, 1H, OH). RMN 13C (100 MHz, DMSO-d6) δ 28,67), 34,65, 40,11, 75,41, 101,05, 124,92, 124,95, 126,44, 126,57, 128,36, 128,53, 128,98, 139,17, 146,10, 167,27, 167,53.
5.3.2 (6 RS)-5,6-Dihydro-4-hydroxy-3-(4-trifluorométhylbenzyl)-6-phényl-2H-pyran-2-one 9
Suivant la procédure générale, sauf en utilisant le α′-chloro-α,α,α-trifluoro-p-xylène (40 μl, 180 mmol), le produit 11 est obtenu (38 mg, 45%) sous la forme d’un solide blanc. Rf : 0,4 (AcOEt/hexane: 50:50). IR, (KBr): 3076, 1712, 1621, 1345, 1069, 827, 692 cm–1. Pf : 105 °C. RMN 1H (400 MHz, DMSO-d6) δ 2,71 (dd, J1 = 17,5 Hz et J2 = 11,5 Hz, 1H, CHH–C–OH), 2,95 (dd, J1 = 17,5 Hz et J2 = 2,6Hz, 1H, CHH–C–OH), 3,50 (s, 2H, CH2–Ar), 5,42 (dd, J1 = 11,5 Hz et J2 = 2,6 Hz, 1H, CH–Ph), 7,02–7,45 (m, 9H, H Ar), 11,05 (s, 1H, OH).
5.3.3 (6 RS)-5,6-Dihydro-4-hydroxy-3-(4-fluorobenzyle)-6-phényl-2H-pyran-2-one 10
Suivant la procédure générale, sauf en utilisant le bromure de p-fluorobenzyle (35 μl, 0,28 mmol), le produit 10 (41 mg, 53%) est obtenu sous la forme d’un solide blanc, Rf: 0,4 (AcOEt/hexane: 50:50). IR, (KBr): 3065, 1788, 1619, 1349, 1068, 872, 631 cm–1. Pf : 173 °C. RMN 1H (400 MHz, DMSO-d6) δ 2,71 (dd, J1 = 17,5 Hz et J2 = 3,1 Hz, 1H, CHH–C–OH), 2,97 (dd, J1 = 17,5 Hz et J2 = 11,5 Hz, 1H, CHH–C–OH), 3,53 (s, 1H, CH2–Ar), 5,43 (dd, J1 = 11,5 Hz et J2 = 3,1 Hz, 1H, CH-Ph), 7,00–7,50 (m, 9H, H Ar), 11,05 (s, 1 H, OH). RMN 13C (100 MHz, DMSO-d6) δ 28,87, 34,64, 75,35, 101,73, 114,57, 114,73, 126,43, 128,50, 129,84, 129,90, 137,13, 139,20, 159,57, 166,71, 167,55.
5.3.4 (6 RS)-5,6-Dihydro-4-hydroxy-3-pentafluorobenzyl-6-phényl-2H-pyran-2 -one 11
Rf:0,1 (AcOEt/hexane: 50:50). IR, (KBr): 3076, 1712, 1621, 1321, 1065, 827, 692 cm–1. Pf: 168 °C. RMN 1H (400 MHz, DMSO-d6) δ 2,72 (dd, J1 = 16,5 Hz et J2 = 3,1 Hz, 1H, CHH–C–OH), 2,94 (dd, J1 = 16,5 Hz et J2 11,2 Hz, 1H, CHH–C–OH), 3,64 (s, 1H, CH2–Ar), 5,38 (dd, J1 = 11,2 Hz et J2 = 3,1 Hz, 1H, CH–Ph), 7,34–7,42 (m, 5H, H Ar), 11,15 (s, 1H, OH). RMN 13C (100 MHz, DMSO-d6) δ 16,57, 34,47, 75,13, 98,24, 114,44, 126,24, 128,18, 128,32, 135,49, 137,47, 138,82, 143,80, 145,74, 166,53, 167,38.
5.3.5 (6 S)-5,6-Dihydro-4-hydroxy-3-benzyl-6-phényl-2H-pyran-2-one 15
À une solution de la lactone 13 (50 mg, 2,6 mmol) dans le diméthylsulfoxyde (2 ml) est ajouté le tert-butoxyde de potassium (32 mg, 2,9 mmol), puis le bromure de benzyle (32 μl, 2,6 mmol). Le mélange réactionnel est agité pendant une nuit à température ambiante puis la saumure (10 ml) est ajoutée. Le mélange est extrait avec l’acétate d’éthyle (2 × 20 ml). La phase organique est séchée sur MgSO4, puis concentrée sous pression réduite. Le brut est chromatographié pour donner le produit d’alkylation 15 (75 mg, 61%) sous la forme d’un solide blanc. Rf : 0,3 (AcOEt/hexane: 50:50). RMN 1H (300 MHz, DMSO-d6) δ 2,71(dd, J1 = 17,l Hz et J2 = 4,0 Hz, 1H,CH–C–OH), 2,97 (dd, J1 = 17,1 Hz et J2 = 11,9 Hz, 1H, CH–C–OH), 3,48 (s, 2H, CH2–Ph), 5,45 (dd, J1 = 11,9 Hz et J2 = 4,0 Hz, 1H, CH–Ph), 7,09–7,50 (m, 10H, H Ar), 11,03 (s, 1H, OH). RMN 13C (75 MHz, DMSO-d6) δ 28,56, 34,62, 75,26, 101,78, 125,44, 126,36, 127,96, 128,14, 128,24, 128,42, 139,17, 140,96, 166,57, 167,57. IR, (KBr) : 3036, 2896, 1796, 1590, 1498, 1292, 1008 cm–1. α = –15.
5.3.6 (6 R)-5,6-Dihydro-4-hydroxy-3-benzyl-6-phényl-2H-pyran-2-one 16
Suivant la procédure générale, sauf en utilisant le bromure de benzyle (32 μl, 2,6 mmol), le produit d’alkylation 16 est obtenu (77 mg, 63%) sous la forme d’un solide blanc. Rf : 0,3 (AcOEt/hexane: 50:50). RMN 1H (300 MHz, DMSO-d6) δ 2,71 (dd, J1 = 17,1 Hz et J2 = 4,0 Hz, 1H, CH–C–OH), 2,97 (dd, J1 = 17,1 Hz et J2 = 11,9 Hz, 1H, CH–C–OH), 3,49 (s, 2H, CH2–Ph), 5,45 (dd, J1 = 11,9 Hz et J2 = 4,0 Hz, 1H, CH–Ph), 7,09–7,50 (m, 10H, H Ar), 11,03 (s, 1H, OH). RMN 13C (75 MHz, DMSO-d6) δ 28,56, 34,62, 75,26, 101,78, 125,44, 126,36, 127,96, 128,14, 128,24, 128,42, 139,17, 140,96, 166,57, 167,57. IR, (KBr): 3036, 2896, 1796, 1590, 1498, 1292, 1008 cm–1. α = 15.