

1 Introduction
Transition-metal-carbonyl complexes, because of their unique spectroscopic properties in the mid-IR range, are now established as useful bioprobes for studies involving high-affinity biological recognition phenomena, such as oestrogen receptorology [1], immunoanalysis [2] and other ligand/protein interactions [3].
These complexes, together with the metal-carbonyl labelled biomolecules, display several characteristic and intense absorption bands in the 1800–2150 cm–1 region, associated to the ν(CO) modes of the carbonyl ligands. The number of ν(CO) bands depends on the local symmetry of the metal carbonyl unit, whereas their position relies on their chemical and molecular environment (organic ligand, solvent [4], pH [5]…).
Several strategies have been exploited to introduce metal-carbonyl groups onto poly-functional biomolecules, including proteins. Most of them rely on electrophilic markers that tag nucleophilic functions (amines, thiols) borne by the biomolecule [6]. One of these strategies takes advantage of the highly electrophilic character of cation [tricarbonyl(1-5-η-cyclohexadienyl)iron](+) that is known to react with O-, N- and S-nucleophiles together with aromatic nucleophiles to yield the tricarbonyl (1-4-η-cyclohexa-1,3-diene) iron 5-substituted adducts [7]. This approach has been followed to label flavonoids [3], and several peptides [8,9]. This latter synthesis was done in water buffered at neutral pH and was shown to occur selectively at the cysteine residue or at the histidine residue in the absence of cysteine.
Compound [tricarbonyl (1-4-η-5-N-pyridiniocyclohexa-1,3-diene) iron] tetrafluoroborate, 1, was shown to be a more convenient alternative to introduce a tricarbonyl (cyclohexa-1,3-diene) iron unit onto proteins in aqueous medium, pyridine being a good leaving group [10,11]. We undertook to study the reaction of 1 with two N-nucleophiles, i.e. Nω-acetyl histamine and n-butylamine and one S-nucleophile, i.e. N-acetyl cysteine (Fig. 1) and characterized the adducts by spectral and elemental analysis.

Synthesis of compounds 2, 3, 4 and 5.
2 Experimental part
2.1 Materials
[Tricarbonyl (1-5-η-cyclohexadienyl)iron][BF4] was synthesized according to an established method [12]. [Tricarbonyl (1-4-η-5-N-pyridiniocyclohexa-1,3-diene) iron][BF4] 1 was prepared from the former complex according to a previously published procedure [13]. Nω-acetylhistamine, n-butylamine and N-acetyl cysteine were purchased from Sigma, Aldrich, and Acros, respectively, and used as received. Reagent-grade acetone (Prolabo) and triethylamine (Aldrich) were used as received. 1H NMR spectra were recorded on a FT-spectrometer (Bruker) operating at 200 MHz. IR spectra were recorded on an MB100 FT-spectrometer (Bomem) equipped with a liquid nitrogen cooled MCT detector. Kinetic measurements were done by injecting the solutions with a micro-syringe in an ultra-microcavity cell in CaF2 (pathlength = 1 mm) from Spectratech.
2.2 Methods
2.2.1 Compound 2
Compound 1 (0.77 g; 2 mmol) in acetone (10 ml) and N-acetyl cysteine (0.326 g ; 2 mmol) in acetone (10 ml) were mixed under nitrogen and the solution was stirred at room temperature. After 3 h, the solution was filtered, then evaporated to dryness under vacuum. The crude solid residue was dissolved in CH2Cl2, washed several times with water, and purified by silica gel column chromatography (eluent: petroleum ether/diethyl ether 1:1). After workup, compound 2 was obtained in 62% yield as a pale yellow powder. Melting point: 120–121 °C.
2.2.2 Compound 3
A solution of compound 1 (0.77 g; 2 mmol) and n-butylamine (0.197 ml; 2 mmol) in acetone (20 ml) was stirred under nitrogen at room temperature for 3 h. The solution was filtered, evaporated to dryness under vacuum and the solid residue was washed with toluene to remove pyridine. Compound 3 was purified by crystallisation in acetone / pentane 2 / 1 mixture and obtained in 85% yield as an off-white powder. Melting point: 141-143°C.
2.2.3 Compound 4
This compound was prepared in an analogous manner as compound 3 and was obtained as a pale yellow solid in 75% yield. Melting point: 127–128 °C.
2.2.4 Compound 5
To a stirred solution of compound 4 (0.918 g; 2 mmol) in dry MeCN (20 ml) was added triethylamine (0.278 ml; 2 mmol). After 30 min, the solution was filtered was evaporated to dryness under vacuum. Purification was performed as described for 2 and compound 5 was crystallised as a pale yellow solid from an acetone/pentane 2:1 mixture.
3 Results
The reaction of 1 with a stoichiometric amount of these molecules at room temperature in acetone yielded after workup off white to pale yellow powders. Spectral and elemental analyses are reported in Table 1.
Spectral and elemental analysis of compounds 2, 3, 4, and 5
| Compound | Yield | 1H NMR (acetone-d6) δ (ppm) | IR (KBr) ν (cm–1) | Anal. |
| 62% |
|
|
|
| 85% |
|
|
|
| 75% |
|
|
|
| 41% |
|
|
|
The IR spectrum of 2, 3, and 4 in KBr pellet displayed two or three intense bands in the 1900–2000 cm–1 region. They were readily assigned to the stretching vibrations of the carbonyl ligands coordinated to iron. According to group theory, two modes of vibration are expected for the C3v symmetry, one symmetrical mode νsym(CO) either called a1 mode and one asymmetrical mode νas(CO) either called e mode. The latter one is known to be degenerate and gives sometimes rise to two bands in the case of solid sampling analysis. Compared to the starting compound 1, no shift of the a1 mode was observed for compound 3 whereas a shift of 3 and 9 cm–1 downwards was noticed for compounds 4 and 2, respectively. The presence of several bands around 1100 cm–1 for compounds 3 and 4 indicated that they were BF4 salts, whereas compound 2, which lacked these bands, was neutral.
1H NMR assignments were done on the basis of a previously published work on imidazolyl [14] and amino ester [15] derivatives and were fully consistent with the formation of 5-S- or 5-N-substituted cyclohexa-1,3-diene adducts. The chemical shifts of the protons belonging to the tricarbonyl cyclohexadiene iron unit were similar for compounds 2, 3, and 4, with protons 6 and 6' being diastereotopic. The organic ligand entities experienced more or less large high-frequency shifts of their protons resonance compared to the parent compounds. For instance, the two protons of the imidazole ring of 4 were shifted by 1.4 ppm (Ha) and 0.74 ppm (Hb) as compared to the parent compound and the two protons of N-acetyl cysteine and n-butylamine adjacent to the heteroatom were shifted by 0.64 and 0.50 ppm, respectively.
Reaction of compound 4 with triethylamine yielded a new compound named 5 as a pale yellow powder. Its IR spectrum still displayed several ν(CO) bands and the position of the a1 mode shifted by 6 cm–1 downwards as compared to compound 4. Moreover, no bands appeared in 1100 cm–1 region, indicating that this compound was neutral. On the basis of previously published results [13,14] and on its 1H NMR spectrum (low frequency shift of H5′ resonance) we assigned compound 5 to the deprotonation product of 4. Interestingly, all the protons’ peaks of the histamine moiety shifted back to the position of the parent compound peaks.
A single signal for proton H5′ indicated that a single stereoisomer was formed by reaction of the N- and S-nucleophiles. On the basis of the small coupling constant between protons H5′ and H6, an exo configuration was assigned to the bioorganic moieties attached to carbon 5. This also indicates that the mechanism of reaction proceeds via dissociation of the labile pyridine group and addition of the nucleophile via the cationic intermediate [(1-5-η-C6H7)Fe(CO)3]+ rather that by nucleophilic substitution at carbon 5 (Fig. 2). To confirm this hypothesis, the reaction of N-acetyl cysteine with 1 was monitored by IR spectroscopy in acetonitrile (Fig. 3).
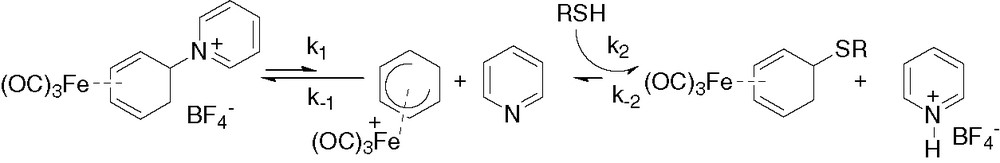
Mechanism of reaction of 1 with N-acetyl cysteine.
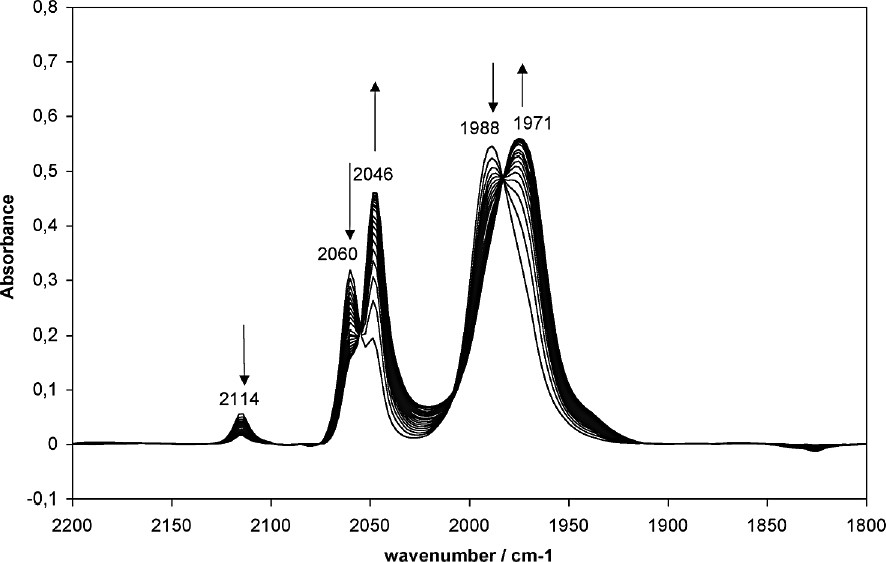
Kinetics of reaction of 1 with N-acetyl cysteine in acetonitrile (concentration = 0.001 M). IR spectra recorded in an ultra-microcavity cell in CaF2 (pathlength = 1 mm) at intervals of 30 s.
In the absence of nucleophile, compound 1 exhibited two ν(CO) bands at 2059 ± 1 cm–1 (a1 mode) and 1989 ± 1 cm–1 (e mode). An additional weaker band at 2114 cm–1 was also observed (Fig. 2) and assigned to the a1 mode of cation [(C6H7)Fe(CO)3]+ resulting from the partial dissociation of pyridine, in agreement with the equilibrium nature of the reaction of this cation with N-nucleophiles [14]. Upon addition of one equivalent of N-acetyl cysteine, the intensity of these three bands was seen to gradually decrease. Concomitantly, another set of two bands at 2046 and 1971 cm–1gradually appeared, corresponding to the formation of compound 3. Curve fitting was applied to the a1 mode band to extract quantitative data. A kinetic analysis of the data revealed that the reaction followed a second order law with a rate constant k of 6 M–1 s–1. The rate-determining step seems therefore to be the addition of the nucleophile to the cationic intermediate, whereas dissociation of pyridine and deprotonation were probably much faster (k1 >> k2). By comparison, the reactions of compound 1 with Nω-acetyl histamine and n-butylamine were too fast to be monitored by IR spectroscopy. The complete replacement of pyridine by Nω-acetyl histamine and n-butylamine is in good agreement with the quantitative nucleophilicity order previously reported [14].
The behaviour of compound 4 in water at pH ranging from 5.3 to 10.8 was studied by IR spectroscopy (Fig. 4).
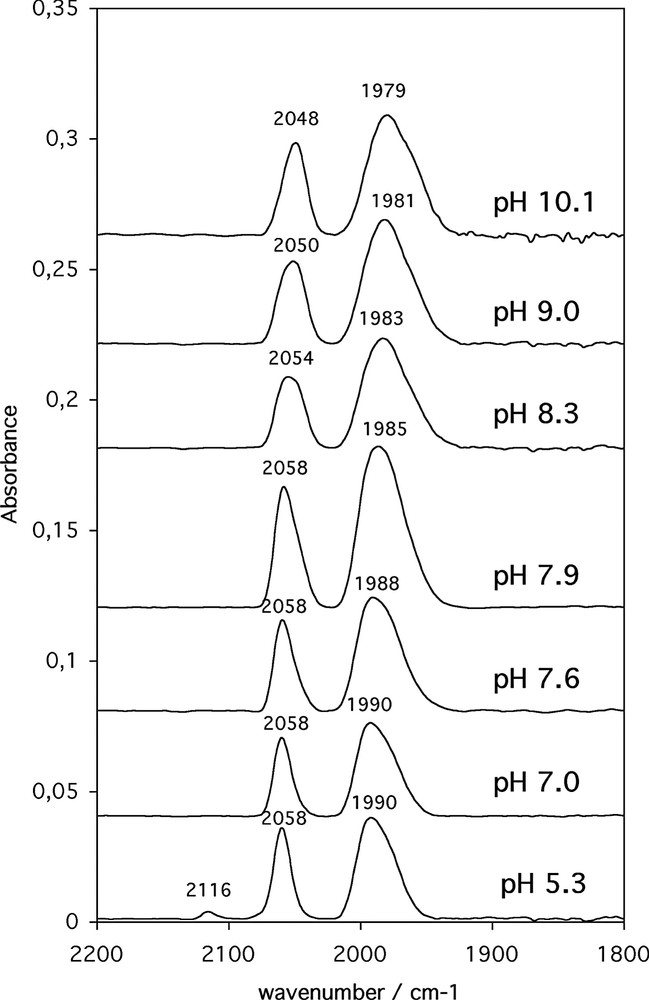
IR spectra of 4 in solution in 0.2-M phosphate buffer pH 5.3 to 7.9 and in 0.1-M carbonate buffer pH 8.3 to 10.1 recorded after deposition of 5 μl of aqueous solutions (concentration = 0.001 M) onto 6 mm diameter nitrocellulose discs and drying in air.
At pH 5.3, a set of two ν(CO) bands was observed at 2058 and 1990 cm–1, readily assigned to the protonated form 4. A weak band at 2116 cm–1 was also observed, indicative of a partial dissociation of N-acetyl histamine. Upon increase of the pH, a shift of the two bands towards the low wavenumbers was observed that was attributed to deprotonation of 4 to yield 5 (2048 and 1979 cm–1) together with the complete disappearance of cation [(C6H7)Fe(CO)3]+. Interestingly, the pKa of this acid/base equilibrium was found roughly around 8.2, as estimated from the set of IR spectra. This value is different from that of the parent compound N-acetyl histamine (pKa = 6.9). Thus, compound 5 is a slightly stronger base than N-acetyl histamine.
In conclusion, we have prepared and characterized cyclohexadiene tricarbonyl iron derivatives of two biologically active molecules, N-acetyl cysteine and N-acetyl histamine and a primary amine by reaction of [tricarbonyl (1-4-η-5-N-pyridiniocyclohexa-1,3-diene) iron] tetrafluoroborate, a η4 precursor of the highly reactive cation [Fe(CO)3(1-5-η-C6H7)](+). Kinetic studies revealed that the reaction of N-acetyl histamine and n-butylamine was much faster than the reaction of N-acetyl cysteine. Finally, the behaviour of the N-acetyl histamine derivative in aqueous medium as monitored by IR spectroscopy revealed that this complex was stable above pH 5 and became deprotonated above pH 8.
Acknowledgements
The CNRS, the French Ministry of Research and the University Mentouri are gratefully acknowledged for financial support.
Vous devez vous connecter pour continuer.
S'authentifier