

1 Introduction
Polyoxometalates (POMs) are structures constituted by arrays of corner- and edge-sharing octahedrally coordinated MO6 units that are packed into an ionic core. The metal M is usually in the d0 or d1 electronic configuration and is designated as an addenda or peripheral element. The most widely studied architectures are Keggin anions, [XM12O40]n– and Dawson anions, [X2M18O62]m–, in which X is the internal element. The most common addenda are represented by WVI, MoVI and VV, and the central heteroatoms usually are P or Si [1–8]. Keeping just with these two basic architectures, the synthetic versatility of POMs makes their number virtually enormous. These early transition metal-oxygen anionic clusters simultaneously exhibit many properties that make them attractive for applications in catalysis, separations, imaging, materials science, and medicine [1–8]. These properties are governed by the atomic composition and the shape of the framework. In this work, we are interested in the electrocatalytic behaviours brought about by addenda substitution as can be exemplified by the presence of vanadium. [9] Specifically, the substitution of vanadium, eventually accompanied by molybdenum, into a W framework of a POM is the focus of this study. The substitution of vanadium into the framework of a tungstic or molybdo-tungstic POM increases the negative charge of the anion and is, therefore, expected to shift the stability of the saturated species to higher pH. Several prominent properties are also recognized for vanadium compounds in general, including polyoxovanadates and vanadium-substituted POMs. For example, in biology, despite the exceedingly complex solution chemistry of vanadate species as a function of pH, the conditions for the efficient oxidation of NADH or phenolic compounds by these cellular components [10,11] and their interactions with proteins were detected and studied [12–14]. Also, a possible role of polyoxovanadates as antitumor and anti-viral agents is being investigated [3]. Synthesis, characterization, electronic structures and catalytic behaviours of vanadium-substituted POMs have generated a vast literature [15–32].
As far as studies in this POM series are concerned, electrochemistry has served several purposes. In addition of mostly analytical and characterization studies and a general survey of Dawson heteropolycompounds [32–35], attention is drawn to the following examples. Examination of the redox potentials of vanadium within POMs substituted in various positions has permitted to reinforce the idea that the first electronation in a Dawson-type species must occur in an equatorial site [19]. Electrochemistry as a function of pH was also associated with ESR in the study of α-[XW9Mo3–xVxO40]n– (X = P and Si, x = 1,2 or 3) concerning electron transfer and electronic properties [36]. Electrocatalytic processes were illustrated both in reductive and in oxidative reactions. The electroreduction of nitric oxide and nitrite in a pH = 2 medium was efficiently carried out by reduced α1-[P2VW17O62]7– [37]. The result was established both in solution and with the catalyst entrapped in slightly quaternized poly(4-vinylpyridine) and immobilized on the electrode surface. The remarkable long-term stability of this assembly is worth noting. Multilayer assemblies consisting of [P2V3W15O62]9– and poly(4-vinylpyridine) partially complexed with osmium bis(2,2′-bipyridine) were also active in the electrocatalytic reduction of bromate and nitrite and the oxidation of ascorbic acid [38]. Regarding oxidation reactions, we have demonstrated that several vanado-tungsto phosphoric heteropolyanions, stable at pH = 7 [39–43], are efficient catalysts for the oxidation of NADH. The mechanism of this process was explored and its rate constant for each active POM was determined. Importantly, the quantitative regeneration of the resulting NAD+ into NADH in the presence of alcohol and alcohol dehydrogenase was achieved.
The present communication develops and expands several new aspects of the electrochemistry and electrocatalytic behaviours of vanadium-substituted POMs. In a selected series of P- and As- Dawson type derivatives, emphasis is put on the stability domain and complete characterization of [P2V2W16O62]8– (P2V2W16 for short) as a representative example, because the redox potential of the vanadium center within this POM is particularly suited for several electrocatalytic processes. Also, several POMs in the series offer the possibility to study and compare mechanistic pathways for both reduction and oxidation catalytic processes merely by modifying the oxidation state of the vanadium center within the same POM. These dual possibilities were illustrated by the oxidation and reduction of nitrogen oxides. Finally, the catalytic oxidation of NAD(P)H into NAD(P)+ has benefited from the synthesis of vanadium-substituted POMs suitable for work in alkaline media.
2 Experimental
2.1 V-substituted POMs
Fig. 1 shows a polyhedral representation of the structure of the basic Dawson polyanion [P2W18O62]6– and the numbering of the metallic atoms according to IUPAC recommendations. This numbering is used in Table 1 which gathers the vanadium-substituted POMs synthesized and the references where their syntheses were described. Only derivatives in the V(V) state are used in this work. Their purity was checked by IR spectroscopy and cyclic voltammetry.
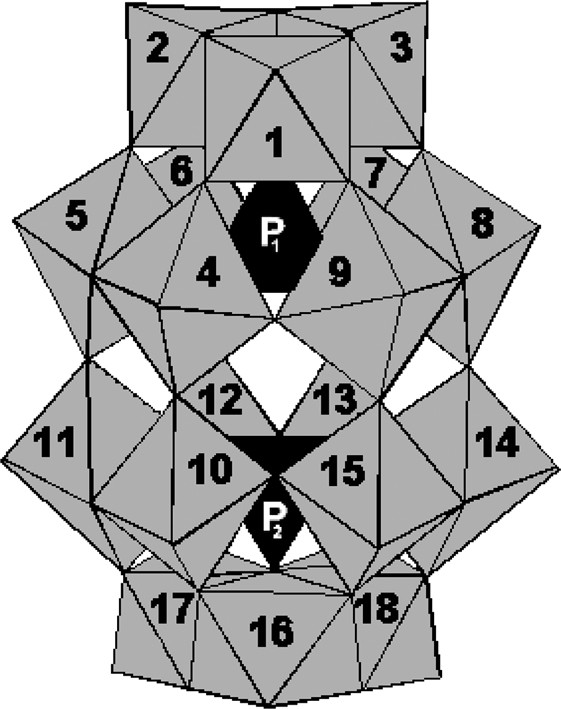
Polyhedral representation of the structure of the basic Dawson polyanion [P2W18O62]6– and the numbering of the metallic atoms according to IUPAC recommendations.
V-substituted Dawson-type POMs synthesized and studied in this work
| Complex | Abbreviation | Reference |
| α1-[As2VW17O62]7– | α1-As2V W17 | 22 |
| α1-[P2VW17O62]7– | α1-P2V W17 | 20 |
| α2-[As2VW17O62]7– | α2-As2V W17 | 22 |
| α2-[P2VW17O62]7– | α2-P2V W17 | 20 |
| 1, 2, 3-[As2Mo2VW15O62]7– | As2Mo2V W15 | This worka |
| 1, 2, 3-[P2V3W15O62]9– | P2V3W15 | 27 |
| 1, 2-[P2V2W16O62]8– | P2V2W16 | 20 |
| 1, 2, 3-[P2MoV2W15O62]8– | P2MoV2W15 | 20 |
a The precursor lacunary complex, 1,2-[As2Mo2W15O61]10–, was prepared by published methods [22], followed by reaction with NaVO3 in HCl medium and precipitation with KCl. After recrystallization,IR spectroscopy and cyclic voltammetry indicated that a pure compound was obtained.
2.2 General Methods and Materials
Pure water was used throughout. It was obtained by passing through a RiOs 8 unit followed by a Millipore-Q Academic purification set. All reagents were of high-purity grade and were used as purchased without further purification. The UV-visible spectra were recorded on a Perkin-Elmer Lambda 19 spectrophotometer on 1.6x10-5 M solutions of the relevant polyanion. Matched 1.000 cm optical path quartz cuvettes were used. The compositions of the various media were as follows: for pH 0: H2SO4; for pH 1 to 3: 0.5 M Na2SO4 + H2SO4 ; for pH 4 and 5: 1 M CH3COONa + CH3COOH ; for pH 7: 0.4 M NaH2PO4 + NaOH; for pH 8; 50mM TRIS + 0.5 M Na2SO4 + H2SO4.
2.3 Electrochemical Experiments
The same media as for UV-visible spectroscopy were used for electrochemistry, but the polyanion concentration was 2 × 10-4 M.
The solutions were thoroughly deaerated with pure argon for at least 30 min and kept under a positive pressure of this gas during the experiments. The source, mounting and polishing of the glassy carbon (GC, Tokai, Japan) electrodes has been described [44]. The glassy carbon samples had a diameter of 3 mm. The electrochemical set-up was an EG & G 273 A driven by a PC with the M270 software. Potentials are quoted against a saturated calomel electrode (SCE). The counter electrode was a platinum gauze of large surface area. All experiments were performed at room temperature.
3 Results and Discussion
3.1 Stability
The stability of the selected complexes was assessed by monitoring their UV–vis spectra between 600 and 200 nm as a function of pH over a period of at least 24h. Table 2 sketches the stability domains for all the derivatives. Fine small dots indicate the domain in which a particular derivative is stable; vertically hachured domains correspond to a decomposition less than or equal to 20%; finally, clear domains indicate a decomposition of between 20 and 40%. Several molecules in their oxidized form remain stable up to pH 7 and further. As expected, the presence of the V-center enlarges the pH stability domain of these mixed addenda heteropolytungstates in comparison with the corresponding monometal main precursors [X2W18O62]6– (X = P or As) and the corresponding Mo-substituted derivatives. A complementary cross-check of this suggested stability was obtained by cyclic voltammetry experiments which usually cover a duration as long as 8 to 10 hours. Perfect reproducibility of the time to time run of the voltammogram for a selected potential scan rate was taken as a complementary stability criterion.
Stability monitored by UV-visible spectroscopy over 24 h, for selected V-substituted Dawson POMs. For further details, see text
3.2 Electrochemistry
P2V2W16 was selected as a representative complex in this study because it can be considered as displaying one of the most complete set of electrochemical behaviors among vanadium-substituted Dawson POMs. Fig. 2A shows the cyclic voltammogram (CV) of this complex in a pH 5 acetate medium. The whole CV is restricted to a potential domain which could be repeatedly cycled without any derivatization of the electrode surface [45] and without any modification of the observed pattern. Provisionally, these experiments underscore again the stability of the reduced species of P2V2W16. The two most positive waves in Fig. 2A feature one-electron processes and are attributed to vanadium centers [19]. Their potential locations are substantially positive to that of processes attributed to W centers. In the following, attention is focused on the electrochemical behaviors of V-centers. Fig. 2B and 2C complete Fig. 2A to give a detailed picture of the evolution of the V-waves as a function of pH. Starting from the pH 5 medium, the two waves move in the positive potential direction as the pH is decreased and, finally, visually merge. However, a clear evolution in peak current intensities and peak widths is still observed between pH 1 and pH 0. A striking evolution of the voltammetric pattern was observed between pH 5 and pH 8 as appears from comparison of Fig. 2A and 2C. The second V-wave is shifted in the negative potential direction and becomes drawn-out and largely irreversible. It is worth noting that the potential location of the first V-wave can be considered to remain fixed in this pH domain. Actually, this independence from pH was observed roughly from pH 4 to higher values. In short, the observed voltammetric pattern can be explained in the following manner: for pH < 4, the first V-wave shows classical potential shifts as a function of acidity and, then, becomes pH-independent for higher pH values; in contrast, the second V-wave experiences much larger displacements, whatever the pH from 0 to 8, a behaviour that induces the coalescene or separation of the two V-waves.
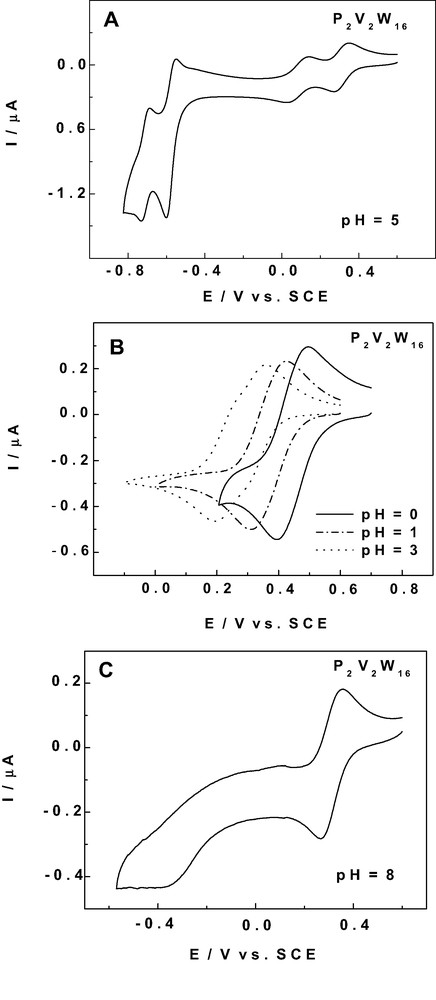
Cyclic voltammograms (CVs) of 2 × 10-4 M P2W16V2 in various media. The scan rate was 2 mV s–1, the working electrode was glassy carbon, and the reference electrode was SCE.
A. pattern showing the two V-waves and two W-waves at pH 5 (1 M CH3COONa + CH3COOH)
B. evolution of the two V-waves as a function of pH. Compositions of the media: pH 0: H2SO4; pH 1 to 3: 0.5 M Na2SO4 + H2SO4.
C. CV showing the two V-waves at pH 8: 50mM TRIS + 0.5 M Na2SO4 + H2SO4
In the following, we are interested, for several of our purposes, in selecting POMs stable in pH domains close to 7 or higher; hence the screening of a series of V-substituted Dawson type complexes and the study of the electrochemical behaviors of V-centers. Table 3 gathers the reduction peak potentials and the apparent formal potentials measured at pH 7 for the V-substituted Dawson POMs studied in this work. Combination of this diversity in the number of V atoms with the presence of As or P as the central heteroatom in these tungstic and molybdo-tungstic structures modulates substantially the apparent formal potentials measured at pH 7 for the first V-wave. The formal redox potentials taken as an average of the cathodic and anodic peak potentials span the range from +569 mV to +122 mV. This leaves considerable flexibility in the choice of POMs for electrocatalytic purposes. It is useful to complete the formal potentials by the comparisons of the actual behaviors of the voltammetric patterns. This should indicate, at least qualitatively, the differences in overall acid-base properties within complexes and their influence on the CVs. Such a comparison is proposed in Fig. 3 between P2MoV2W15 and P2V3W15 which completes Fig. 2C. The pattern for P2MoV2W15 at pH 8 in Fig. 3 is very closely the same as that of P2V2W16 in Fig. 2C, albeit for a very slight difference in formal potentials. It is likely that the CVs are dominated by the V centers, despite the presence of a Mo atom. In Fig. 3, P2MoV2W15 was studied at pH 8, but P2V3W15 at pH 7 to highlight the subtle difference in acid-base properties between these complexes. The CV for the latter complex displays a drawn-out first wave with a broad reoxidation counterpart. The second part is composed of two closely-spaced cathodic waves, which are irreversible on potential reversal. These waves are likely to feature the reduction processes of the two remaining V-centers. In summary, as a qualitative outcome of the comparisons, it is worth emphasizing that the beneficial influence expected from accumulation of V-centers in P2V3W15 as compared to P2V2W16 and P2MoV2W15 might be counterbalanced by the qualitatively poorer acid-base and electron transfer abilities of the former complex.
V-reduction peak potential Epc1 and apparent formal potentials E°′ (defined as (Epc1 + Epa1)/2) measured in a pH 7 medium by cyclic voltammetry for a selection of V-substituted Dawson POMs; scan rate of 2 mV s–1; reference electrode: SCE; working electrode: glassy carbon
| Complex | Epc1/mV | E°’/mV |
| α1-As2V W17 | 531 | 569 |
| α1-P2V W17 | 453 | 504 |
| α2-As2V W17 | 404 | 446 |
| α2-P2V W17 | 377 | 414 |
| As2Mo2V W15 | 455 | 489 |
| P2V3W15 | 59 | 122 |
| P2V2W16 | 255 | 298 |
| P2MoV2W15 | 267 | 309 |
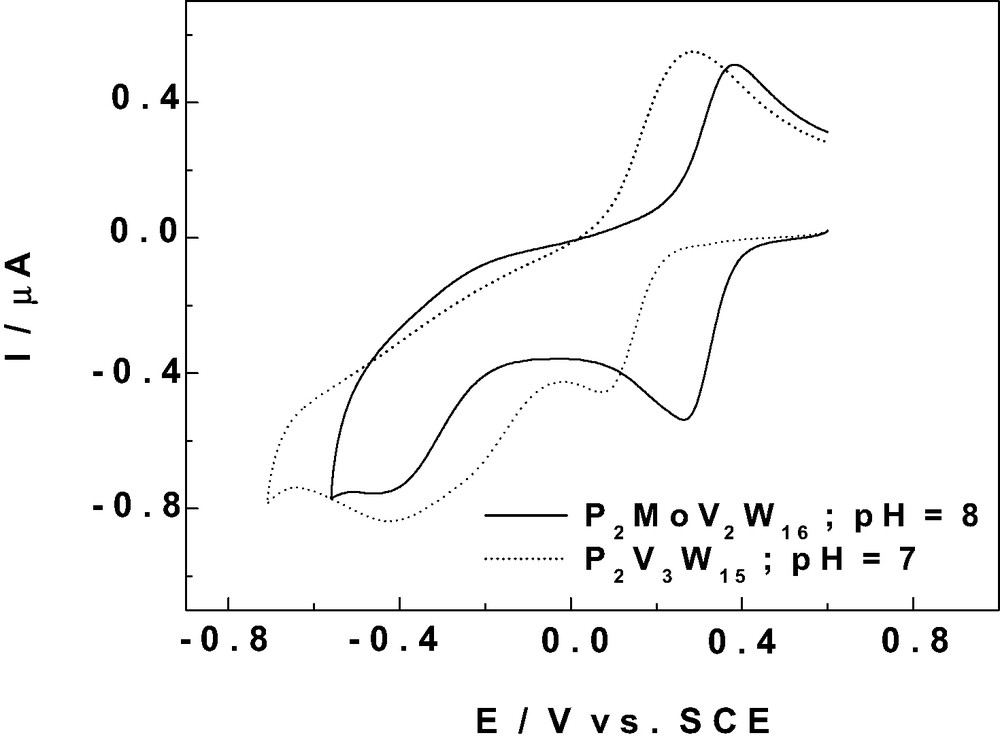
Comparison of the cyclic voltammograms (CVs) of 2 × 10-4 M P2MoW15V2 (in pH 8: 50mM TRIS + 0.5 M Na2SO4 + H2SO4) and 2 × 10-4 M P2V3W15 (in pH 7: 0.4 M NaH2PO4 + NaOH). The scan rate was 2 mV s–1, the working electrode was glassy carbon, and the reference electrode was SCE. For further details, see text.
3.3 Electrocatalysis
3.3.1 Electrocatalysis of the reduction of nitrite: an example under continuous improvement
Catalytic reduction of the NOx and particularly of nitrite by reduced POMs has become a classical test for their electrocatalytic abilities since the pioneering works in this area [46–50]. Examples concerning specifically V-substituted POMs were also studied [37,38]. General aspects of such electrocatalytic processes were discussed recently [51]. The favourable ability of the V-containing POMs is illustrated with one example. Fig. 4 shows the current enhancement accompanying the addition of NaNO2 to a pH 1 solution [52] of P2V2W16.
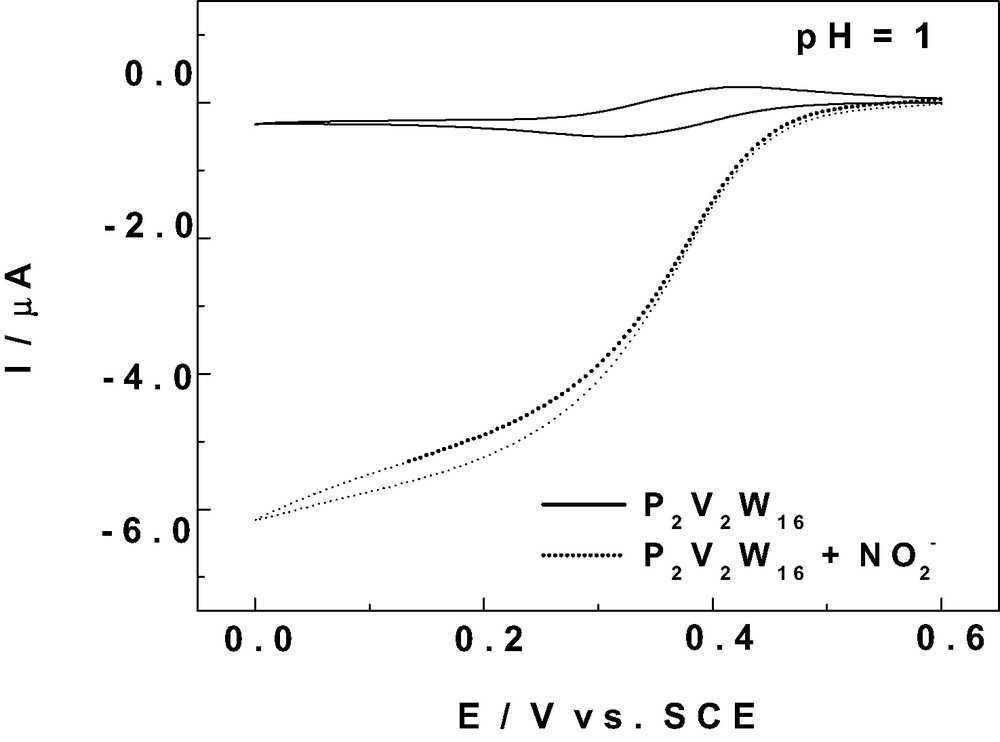
Cyclic voltammetry study of the electrocatalytic reduction of 2 × 10-2 M NaNO2 (γ = 100) with a 2 × 10-4 M solution of P2V2W16 in a pH = 1 medium (0.5 M Na2SO4 + H2SO4). The scan rate was 2 mV s–1, the working electrode was glassy carbon, and the reference electrode was SCE.
The pH 1 value was selected because it ensures a fairly complete merging of the two V-waves of this complex. The current intensity increase is observed readily at the reduction potential of the V-centers for a modest increase of γ values (γ is the excess parameter defined as γ = C° (NOx) / C° (POM)). In this potential domain, no direct reduction is observed for NO or HNO2 present in the solution [37,49]. Worth of notice are the high efficiency of the electrocatalysis and the fairly positive potential at which it takes place. The main incentive to show Fig. 4 is to stress the possibility that an appropriate selection of POMs could be performed to obtain a continuous shift in the positive direction of the potential at which such electrocatalysis is observed. The limit is the possibility that nitrite be oxidized into nitrate by the POM instead of undergoing an electrocatalytic reduction process. This point is considered in the following.
3.3.2 An example of mediated oxidation process: oxidation of nitrite
Putatively demonstrative POMs were selected by taking into account their formal potentials in Table 3. Preliminary experiments were performed by simply mixing one selected POM and nitrite in a pH 7 medium and observing eventual colour changes as a function of time. Then, more quantitative experiments were realized by UV-vis spectroscopy. A representative example is illustrated by the addition of a fixed excess of nitrite (defined by γ, vide supra) to a pH = 7 solution of α1-P2VVW17. The evolution of the mixture was monitored by UV-vis spectroscopy between 1300 nm and 400 nm. Fig. 5 shows a set of spectra. Starting with the neat POM (γ = 0), addition of nitrite (γ = 100) results in the reduction of the V-center and the gradual development of the known spectrum of the reduced POM. This spectrum is the same as that of the heteropolyblue prepared by electrochemical reduction. Interestingly, it is fairly different from that of the oxidized form. A sharp isosbestic point indicates a neat conversion of one form into the other. Complete kinetic study of this process is beyond the scope of the present work. However, the set of spectra in Fig. 5 can be exploited to calculate a pseudo-first order rate constant k′ = 1.1 × 10-3 s-1 under the assumption that the rate is first order in POM concentration. Table 4 gathers the main observations with the POMs studied in this work. Despite their mostly qualitative character, the results suggest two main parameters to be operative in this issue, in addition of the necessary stability of the two relevant redox states of the POM: the formal potential of the POMs and the pH of the solution. Specifically, α1-As2VVW17 is chosen as a representative example. The influence of pH might be twofold: possibly shifting the formal potential(s) of the V-wave(s) in the cases of several complexes like P2V2W16; and also fixing the equilibrium state of nitrite in solution1, and hence the nature of the species to be actually oxidized by the POM. Then, it is conceivable that the oxidation of HNO2 (and eventually NO) be difficult at pH 1. The first waves of α2-As2V W17 and α1-P2VW17 are located at less positive potentials than the corresponding wave of α1-As2VW17 at pH 2. Hence, the slower nitrite oxidation kinetics observed for the two former complexes. Finally, still less positive potentials measured for P2MoV2W15 and P2V2W16, respectively, justify that nearly no oxidation of nitrite by these POMs be obtained at pH 7. All these considerations lead to the conclusion that a redox type reaction is operative in the oxidation of nitrite by the POMs gathered in Table 4. Even though electrochemical regeneration of the catalytically active forms can be easily envisioned in favourable cases to complete an electrocatalytic cycle, the present examples, due to their relative slowness, are clearly better handled by UV-vis spectroscopy for their characterization.
3.3.3 Electrocatalytic oxidation of NAD(P)H in alkaline media
Previous work from our group has established the efficient oxidation of NADH by several vanado-tungsto phosphoric heteropolyanions, stable at pH = 7 [39–43]. The series of V-substituted derivatives selected in this work contains several POMs stable in their oxidized and reduced forms in alkaline media, thus guaranteeing the stability of the species generated during the catalytic process. The present preliminary study is devoted to the electrocatalytic oxidation of NADPH, a substrate closely related to NADH, with the same importance in vivo and in vitro. Fig. 6A shows the current enhancements accompanying the addition of increasing amounts of NADPH to a pH 8 solution of P2V2W16. The current intensity increase is observed readily at the reduction potential of the first V- center. It is worth noting the high efficiency of the electrocatalysis, even though modest γ values were used (γ is the excess parameter defined here as γ = C° (NADPH)/C° (POM)). To characterize the process itself, the catalytic efficiency (CAT) is defined as follows: CAT = 100 × [I (POM + NADPH) – Id (POM)] / Id (POM), where I (POM + NADPH) is the current for the reduction of the POM in the presence of NADPH and Id is the corresponding diffusion current for the POM alone. In the present experiments, CAT values of 403% and 730% were calculated for γ = 10 and γ = 20 respectively at + 308 mV vs SCE. Fig. 6B shows the direct NADPH oxidation wave on the glassy carbon electrode surface and the rising section of the catalytic wave for γ = 20 in the same pH 8 medium. It is clear from this Figure that no direct oxidation of NADPH is observed in this potential domain explored in Fig. 6A. Furthermore, the substantial improvement in potential brought about by the catalysis for the oxidation of NADPH is obvious. Double potential step chronocoulometry experiments [41] performed between 0 and + 400 mV vs SCE had allowed the value of the second order rate constant to be evaluated: k = 3.5 × 103 M-1 s-1. The present experiments were performed in deaerated solution. However, we have checked that an inert atmosphere was not necessary, because the VV and VIV forms of P2V2W16 are insensitive to the presence of dioxygen. This remark is important in that it opens the way for working in conditions close to those of biological environments, in contrast with most mediators, especially quinonic mediators proposed in the literature [52].
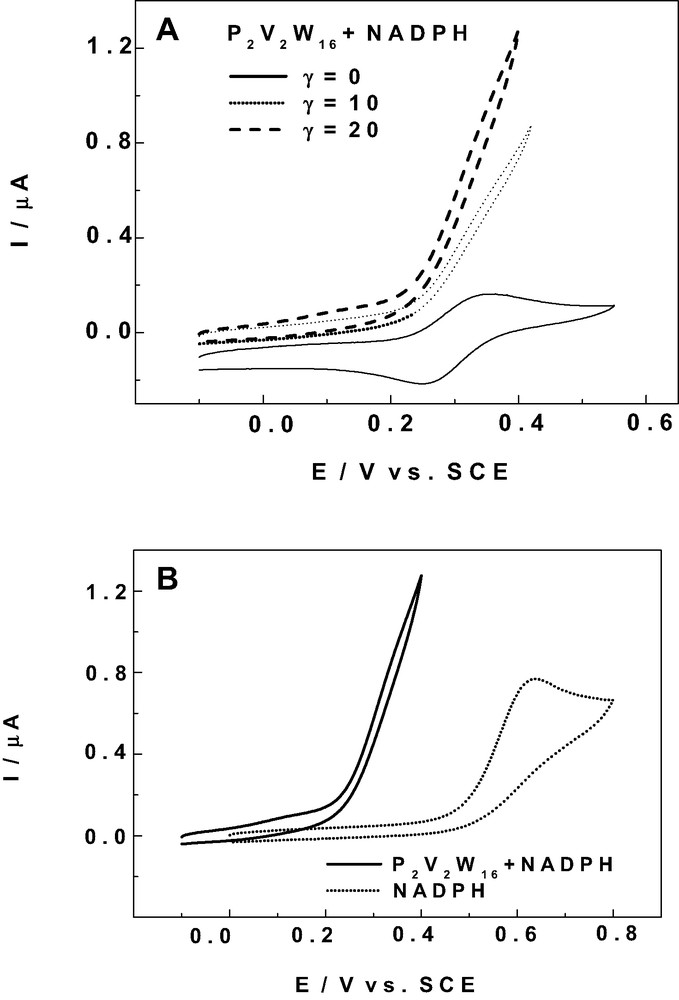
Cyclic voltammograms for the electrocatalytic oxidation of NADPH, by 2 × 10-4 M P2W16V2, in pH 8 buffer (50mM TRIS + 0.5 M Na2SO4 + H2SO4). The scan rate was 2 mV s–1, the working electrode was glassy carbon, the reference electrode was SCE, (A) The excess parameter values for NADPH were γ = 0, γ = 10 and γ = 20 respectively; (B) Comparison of the catalytic process (γ = 20) by P2V2W16 with the direct oxidation of NADPH on the glassy carbon electrode.
4 Concluding remarks
The Dawson type V-substituted tungsto- and molybdo-tungsto- diarsenates and diphosphates synthesized and studied in this work display a series of interesting properties: (i) the presence of vanadium atom(s) is beneficial in driving the pH stability domains of the substituted POMs towards high values, a feature important in several catalytic and electrocatalytic processes; (ii) the VIV forms are more stable than VV forms in high pH media, a feature usually shared by reduced vs oxidized POMs; (iii) combination of various atomic compositions has permitted the selection and synthesis of a collection of POMs with the V-wave(s) locations spanning a large potential domain. Most of these properties are illustrated in the electrochemistry of P2V2W16 chosen as a representative example. One feature worth of notice is the high sensitivity of the second V-wave to pH variations when compared to the first one.
Finally, the V-center(s) might be considered either as a reducing or an oxidizing center. Even though a simple definition of a bicatalyst might imply the presence of two different metallic centers within the same molecule, it is tempting to extend that concept to the oxidized and reduced V-center(s) within the present POMs. As a matter of fact, the results of this study establish the ability of an appropriate oxidation state of the V-center to act as a reducing or an oxidizing center toward the same substrate, provided the pH of the media were fixed accordingly.
Work in progress will take advantage of these dual possibilities.
Acknowledgements
This work was supported by the University Paris Sud XI and the CNRS (UMR 8000). Dr Yu Wei Lu is thanked for his help in the calculation of a kinetic rate constant.
1 At pH = 1, the actual active species should be HNO2 and/or NO. In fact, the following sequence is known: HNO2 ⇌ H+ + NO2– pKa = 3.3 at 18 °C and HNO2 disproportionates in fairly acidic solution: 3 HNO2 ⇌ HNO3 + 2 NO + H2O. The rate of this reaction is known to be low.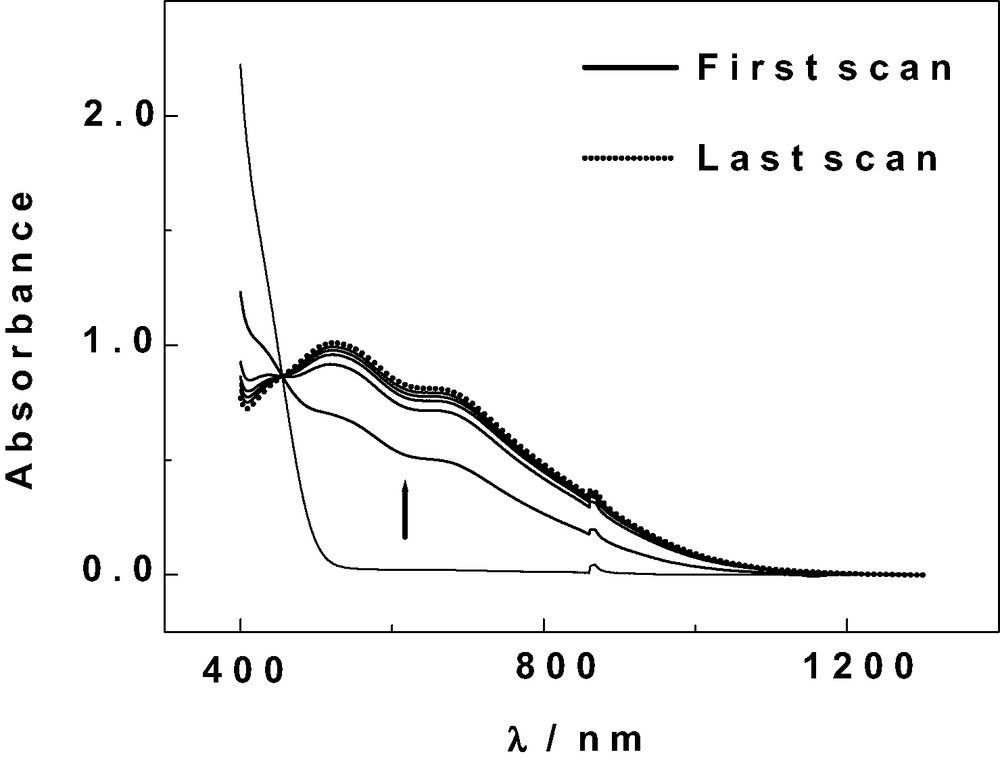
| Complex | pH | Qualitative kinetics | Isosbestic point on the spectra | Comments |
| α1-As2VV W17 | 1 | slow | yes | Incomplete after 7 h |
| 2 | fast | yes | Complete in a few minutes | |
| 5 | fast | yes | Complete in a few minutes | |
| 7 | very fast | yes | Complete in a few minutes | |
| α2-As2VV W17 | 7 | slow | no | Incomplete after 7 h |
| α1-P2VV W17 | 2 | slow | yes | Incomplete after 7 h |
| 5 | fast | yes | Complete in a few minutes | |
| 7 | very fast | yes | The excess parameter γ must be decreased for a good observation of the isosbestic point | |
| P2MoV2W15 | 7 | Very small conversion ratio | ||
| P2V2W16 | 7 | Negligible conversion ratio |