

1 Introduction
The design of clusters containing a large number of metal centers represents an intellectually stimulating challenge for the chemist since the nano-scale chemistry are situated at the boundary of molecular and condensed matter and envelops diverse fields such as biochemical processes, [1] catalysis [2–4] and material science [5,6]. In this context, cyclic transition metal clusters based on the {M2S2O2} building block constitute a fascinating emerging class of compounds [7]. The cyclic architecture of the cluster delimits a central cavity which presents a significant cationic character at the origin of striking properties related with supramolecular [8] and host–guest chemistry [9,10]. Anionic reagents such as phosphate or dicarboxylate ions have been inserted in the cavity by replacing the water molecules which initially line the cavity [11]. The substitution leads to size-controlled molecular wheels, more or less distorted and containing from eight [12] to 16 [13] metallic atoms. NMR studies in solution have revealed that strong dynamic synergistic effects between the guest and the host exist, reflecting the high flexibility of the hybrid architecture [14]. In a continuing project, we systematically investigate the host–guest interactions in such hybrid molecular materials, the goal being the understanding of the rules which govern the adaptability, the flexibility, the dynamic and the exchange properties of the encapsulated templates [15], because such compounds could be used for the inclusion of specific anionic substrates for transport or extraction processes. Herein, we report on the synthesis, the structural characterization and the behavior in solution of two cyclic hybrid molecular compounds [Mo12S12O12(OH)12(C4H4O4)2]4– (1, noted [Mo12-succ2]4–) and [Mo12S12O12(OH)12(H2O)4(C4H2O4)]2– (2, noted [Mo12-fum]2–), which consist of a Mo12-ring templated by a four carbon linear dicarboxylate ion, i.e. fumarate and succinate ions, respectively.
2 Experimental
2.1 Synthesis
2.1.1 Rb3(NMe4)[Mo12-succ2]·25 H2O (1)
The crude precursor K2I2Mo10S10O10(OH)10(H2O)5]·15 H2O·ε NMe4I [11] (1 g; 0.41 mmol) was dissolved in 80 ml of water at 50 °C. Succinic acid (0.1 g; 0.8 mmol) was dissolved in 10 ml of water, basified by 1.6 ml of 1 mol l–1 KOH solution. The resultant potassium succinate solution was added to the previous one for giving an orange solution (about pH 5). The mixture was kept at 50 °C for 45 min and rubidium chloride (2.15 g; 17.7 mmol) is added. A yellow cloudy precipitate was removed by filtration and the resulting filtrate allowed to stand in air for crystallization. After several days, orange crystals suitable for X-ray diffraction are obtained, washed by water and dried in air. Yield 42%. Anal. Calc. (found) for 1: Mo 39.09 (39.53), S 13.06 (12.90). EDX atomic ratio calcd. (found): Mo/S, 1 (0.98); Mo/Rb, 4 (3.90); S/Rb, 4 (3.96). IR/cm–1 (KBr pellet):1543(s), 1484(m), 1453(m), 1398(m), 939(s), 496(s), NMR 1H in DMSO-d6 per ppm: –0.70, 2.30, 3.04, 10.34.
2.1.2 Rb1,5K0.5[Mo12-fum]·19 H2O (2)
The crude precursor K2I2Mo10S10O10(OH)10(H2O)5]·15 H2O·ε NMe4I [11] (1 g; 0.41 mmol) was dissolved in 60 ml of water at 50 °C. Fumaric acid (0.076 g; 0.60 mmol) was dissolved in 10 ml of water and 1.2 ml of 1 mol l–1 KOH solution. The resulting solution was added to the previous solution for giving a clear orange solution at pH about 4.5. A first fraction of the target compound was obtained by the addition of RbCl (0.205 g; 1.7 mmol) to the solution (yield 32%). Single crystals suitable for the X-ray diffraction study were obtained from the filtrate which was allowed to stand for 1 week (yield 30%). Anal. Calc. (found) for 1: Rb 4.91 (4.79), K 0.75 (0.85) Mo 44.14 (44.63), S 14.75 (14.66), C 1.84 (2.23). EDX atomic ratio calcd. (found): Mo/S, 1 (1.13); Mo/Rb, 8 (7.97); S/Rb, 8 (8.3). IR/cm–1 (KBr pellet):1558(s), 1387(m), 962(m), 521(m). NMR 1H in DMSO-d6 per ppm: 2.99, 10.31, 3.04, 10.34.
2.1.3 Li4[Mo12_succ2]·n H2O and Li2[Mo12-fum]·n H2O
The two lithium salts were prepared from the corresponding potassium salt K4[Mo12_succ2]·nH2O and K2[Mo12-fum]·n H2O, previously dissolved in water (about 0.4 g in 30 ml of water). Each solution was passed through a cation exchanger resin in the Li+ form (Dowex 50WX2). The resultant solutions were evaporated until dryness, and the purity of the lithium salt checked by routine methods (1H NMR, IR, EDX).
The potassium salts of 1 and 2 were prepared as the procedures previously described for the rubidium salt (see below). Instead RbCl, potassium chloride (4 g; 53.6 mmol) was added for the precipitation of the corresponding potassium salts. The crystalline solids of 1 and 2, as potassium salts have been characterized by routine methods (1H NMR, IR, EDX).
2.2 Physical methods
Elemental analysis was performed by the ‘Laboratoire central d’analyse du CNRS’ (Solaize, France). Water content was determined by thermal gravimetric analysis (tga7, Perkin–Elmer).
Infrared spectra were recorded on an Magna 550 Nicolet spectrophotometer, using KBr pellet.
NMR measurements were performed on a Brüker Avance 300 spectrometer, operating at 300 MHz in 5 mm tubes. Chemical shifts were referenced to the external usual TMS standard. The variable temperature 1H NMR studies were performed in CD3CN solution, 0.1 mol l–1 of lithium salt, Li4[Mo12_succ2]·n H2O or Li2[Mo12-fum]·n H2O, respectively.
2.2.1 X-ray crystallography
A single crystal of 1 was mounted in a 0.3 mm diameter Lindeman tube while a single crystal of 2 was stabilized in liquid glycerin (paratone). Intensity data were collected at room temperature (298 K) for 1 and 100 K for 2 with a Brüker X8, Kappa CCD three-circle diffractometer equipped using the Mo Kα monochromatized wavelength (λ = 0.71073 Å). An empirical absorption correction was applied using the SADABS program based on the method of Blessing [16,17]. The structures were solved by direct methods and refined by full-matrix least-squares using the SHELX-TL package [18]. Crystallographic data for 1 and 2 are reported in Table 1, selected bond distances are given in Table 2. Some Rb+ cations and some oxygen atoms of water molecules were found disordered. All the disordered atoms and water of crystallization were refined isotropically, the other atoms were refined anisotropically. Hydrogen atoms of the alkyl chains of the succinate and fumarate ions were located with geometrical restraints in the riding mode. Lattice water found by single crystal X-ray diffraction method slightly disagrees with those found by TGA method for 1 and 2 crystals. Such a difference is currently encountered for those ionic compounds: water molecules are often highly disordered and crystals decompose in air with loss of water.
Crystal data for 1 and 2
| 1 | 2 | |
| Formula | C12H54Mo12NO43Rb3S12 | C4H74K0.50Mo12O58Rb1.50S12 |
| M (g mol–1) | 2734.39 | 2692.97 |
| T (K) | 298(2) | 100(2) |
| Crystal size (mm) | 0.32 × 0.16 × 0.16 | 0.26 × 0.16 × 0.06 |
| Crystal system | Orthorhombic | Triclinic |
| Space group | Immm | P |
| a (Å) | 18.6607(13) | 9.203(2) |
| b (Å) | 11.0184(6) | 14.055(4) |
| c (Å) | 21.4067(12) | 16.272(4) |
| α (°) | 90 | 106.15(1) |
| β (°) | 90 | 97.91(1) |
| γ (°) | 90 | 99.53(1) |
| V (Å3) | 4401.5(5) | 1956.1(9) |
| Z | 2 | 1 |
| ρcalc (g cm–3) | 2.032 | 2.321 |
| μ (Mo Kα) (cm–1) | 3.648 | 3.222 |
| λ (Mo Kα) (Å) | 0.71073 | 0.71073 |
| θ Range (°) | 1.45–29.98 | 1.33–40.95 |
| Data collected | 25,847 | 64,747 |
| Unique data | 3490 | 24,788 |
| Unique data I > 2σ(I) | 2315 | 15,921 |
| Number of parameters | 128 | 424 |
| R(F)a | 0.0644 | 0.0372 |
| Rw(F2)b | 0.0893 | 0.0749 |
| Goodness-of-fit | 1.092 | 1.026 |
Selected bond lengths
| For (1) | ||
| Mo1 | O1 | 1.710(6) |
| O4 | 2.103(5) | |
| O4 | 2.103(5) | |
| S1 | 2.318(2) | |
| S1 | 2.318(2) | |
| O6 | 2.336(9) | |
| Mo2 | 2.821(1) | |
| Mo1 | 3.385(1) | |
| Mo2 | O2 | 1.694(9) |
| O5 | 2.101(5) | |
| O5 | 2.101(5) | |
| S1 | 2.325(2) | |
| S1 | 2.325(2) | |
| O7 | 2.325(7) | |
| Mo1 | 2.821(1) | |
| Mo3 | 3.281(1) | |
| Mo3 | O3 | 1.687(8) |
| O5 | 2.088(4) | |
| O5 | 2.088(4) | |
| S2 | 2.323(2) | |
| S2 | 2.323(2) | |
| O7 | 2.579(7) | |
| Mo3 | 2.841(1) | |
| Mo2 | 3.281(1) | |
| C1 | O7 | 1.225(2) |
| O6 | 1.262(2) | |
| C2 | 1.597(2) | |
| C2 | C2 | 1.483(2) |
| For (2) | ||
| Mo1 | O1 | 1.690(1) |
| O7 | 2.099(1) | |
| O8 | 2.125(1) | |
| S1 | 2.323(1) | |
| S2 | 2.330(2) | |
| O13 | 2.345(1) | |
| Mo2 | 2.831(1) | |
| Mo6 | 3.383(1) | |
| Mo2 | O2 | 1.695(1) |
| O10 | 2.096(2) | |
| O9 | 2.116(1) | |
| S2 | 2.318(1) | |
| S1 | 2.326(2) | |
| O14 | 2.541(2) | |
| Mo1 | 2.831(1) | |
| Mo3 | 3.329(2) | |
| Mo3 | O3 | 1.673(1) |
| O10 | 2.085(1) | |
| O9 | 2.118(2) | |
| S3 | 2.315(1) | |
| S4 | 2.329(2) | |
| O14 | 2.645(1) | |
| Mo4 | 2.834(2) | |
| Mo2 | 3.329(2) | |
| Mo4 | O4 | 1.682(1) |
| O11 | 2.067(1) | |
| O12 | 2.109(1) | |
| S3 | 2.325(2) | |
| S4 | 2.331(1) | |
| O15 | 2.606(1) | |
| Mo3 | 2.834(2) | |
| Mo5 | 3.316(1) | |
| O3 | 3.498(2) | |
| Mo5 | O5 | 1.683(1) |
| O11 | 2.079(1) | |
| O12 | 2.100(1) | |
| S6 | 2.328(2) | |
| S5 | 2.332(1) | |
| O15 | 2.583(2) | |
| Mo6 | 2.855(2) | |
| Mo4 | 3.316(1) | |
| Mo6 | O6 | 1.690(1) |
| O7 | 2.109(1) | |
| O8 | 2.136(2) | |
| O16 | 2.271(2) | |
| S5 | 2.325(1) | |
| S6 | 2.341(1) | |
| Mo5 | 2.855(2) | |
| Mo1 | 3.383(2) | |
| C1 | O13 | 1.262(1) |
| O16 | 1.276(1) | |
| C2 | 1.494(1) | |
| C2 | C2 | 1.309(1) |
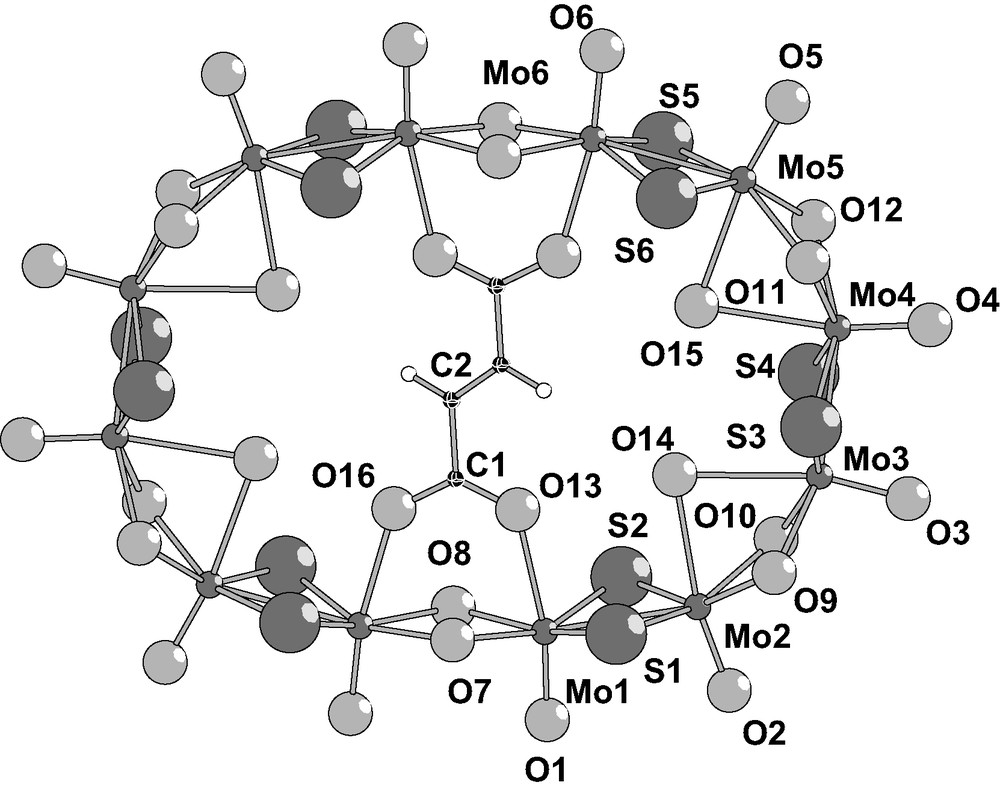
2.2.2 Structure of the anions
In Figs. 1 and 2 are depicted the molecular structure of the [Mo12-succ2]4– (1) and [Mo12-fum]2– (2) anions. Crystal data and selected bond distances are given in Tables 1 and 2, respectively. The two [Mo12-fum]2– and [Mo12-succ2]4– molecular architectures consist of an inorganic cyclic neutral skeleton {Mo12S12O12(OH)12}, encapsulating two succinate ion for 1 and one fumarate ion for 2, respectively. The inorganic wheel results from {Mo2O2S2}2+ connections through double hydroxo bridges, giving non-bonding MoLMo contacts (3.282(1)–3.383(1) Å) while sulfur atoms within the building blocks spans the MoLMo bonding contacts (2.821(1)–2.855(2) Å).

Molecular structure of [Mo12S12O12(OH)12(C4H4O4)2]4– (1).
Molecular structure of [Mo12S12O12(OH)12(OH2)4(C4H2O4)]2– (2).
2.2.3 [Mo12S12O12(OH)12(H2O)4(C4H4O4)2]4– (1)
The Mo12-inorganic wheel encapsulates two succinate ions which are symmetrically arranged in the open-cavity (see Fig. 1). The four carboxylato groups exhibit the same asymmetric binding mode, each group being opposite to a single {Mo2O2S2}. The O6 oxygen atom exhibits only one bonding contact with the Mo1 atom, while the O7 atom bridges the Mo2 and Mo3 atoms. The 12 molybdenum exhibit an octahedral arrangement and are arranged in a C2v idealized local symmetry.
2.2.4 [Mo12S12O12(OH)12(OH2)4(C4H2O4)]2– (2)
The structure reveals the presence of one encapsulated fumarate ion [H2C4O4]2– in the Mo12-ring (Fig. 2). The organic template is nearly located at the center of the elliptically distorted ring (C2h idealized molecular symmetry) and connected through the two symmetric carboxylato groups. Four molybdenum atoms are involved in a bonding contact with the organic guest, while the coordination sphere of the other eight is filled by four inner water molecules (O14 and O15) which are doubly bonded to two adjacent molybdenum atoms. The carboxylato groups bridge two adjacent molybdenum atoms (Mo1 and Mo6), belonging to two different {Mo2O2S2}. Such a mode of connection differs of those previously encountered, where the carboxylato groups are directly connected to the two adjacent molybdenum atom of the same {Mo2O2S2} unit [14]. Such a result is attributed to steric constraints induced by the rigidity of the central C=C double bond of the guest, which imposes the conformational arrangement of the inorganic host around the rigid template.
2.2.5 1H NMR in D2O
Besides the signal of the solvent (presence of H2O), the spectrum of [Mo12-fum]2– (not shown) exhibits a sharp resonance at 3.02 ppm corresponding to the two equivalent protons attached to the encapsulated fumarate ion. The uncoordinated fumarate is observed at 6.4 ppm. The 1H NMR spectrum of the [Mo12-succ2]4– (Fig. 3) reveals more complicated, since three resonances corresponding to three different environments for the succinate ions are observed. At 295 K, the broad peak at 2.2 ppm is attributed to the uncoordinated succinate ions, while the two shielded resonances at –0.01 and –0.66 ppm correspond to encapsulated species. The presence of uncoordinated succinate ions reveals that succinate ion exchange occurs in the ring, and the simplest equilibrium (Eq. 1) can be postulated.
| [Mo12-Succ2]4– ⇌ [Mo12–succ]2– + succ2– | (1) |

Selected variable-temperature 1H NMR spectra of [Mo12S12O12(OH)12(C4H4O4)2]4– (1) in D2O.
The intensity and the broadness of the –0.01 ppm line should argues for its attribution to the mono-succinate species. At upper temperature than 300 K, this line (–0.01 ppm) and that at 2.2 ppm exhibits nearly equal intensities according to the equilibrium 1, while the broadness of the –0.01 ppm line (Δν1/2 = 50 Hz) would reflect the possibility of the remaining encapsulated succinate ion to move inside the half-empty cavity of Mo12-ring. The remaining line at –0.66 ppm would be then attributed to the bis succinato-containing species [Mo12-succ2]4–, involved in the equilibrium 1. In an aprotic solvent as CD3CN, the equilibrium 1 reveals totally cancelled since only one line is observed at –0.65 ppm, corresponding to the bis succinato compound [Mo12-succ2]4–. The decrease of the temperature from 310 to 275 K changes drastically the distribution in the species as shown by the variable temperature 1H NMR experiments presented in Fig. 3. A second equilibrium (Eq. 2) must be considered necessarily, involving a second exchange process of the succinate ion from the mono-succinato compound [Mo12-succ]2–.
| [Mo12-Succ]2– ⇌ [Mo12] + succ2– | (2) |
The equilibrium 2 gives an additional species, i.e. a templated-free Mo12-ring, noted [Mo12]. The concentration of overall species can be calculated from the initial concentration in [Mo12-succ2]4– (C = 3.85 × 10–3 mol l–1), the intensities of the 1H NMR lines, the mass balance in succinate and in Mo12-species. The results are graphically shown in Fig. 4a and reveal that as temperature decreases, the concentration in [Mo12-succ2]4– strongly decreases for giving uncoordinated succinate ions and succinate-free Mo12 compound. As a consequence of the successive equilibria, concentrations of the intermediary compound [Mo12-succ]2– do not vary significantly. Then, the equilibrium constants K1 and K2 could be calculated in the 310–273 K temperature range, according to the Eqs. 3 and 4, respectively.

(a) Variation of the distribution in [Mo12-succ2]4– (Δ), [Mo12-succ]2– (□), [Mo12] (◊), [succ]2– (○) with temperature; (b) Van’t Hoff linear plots lnKi = f(1/T) with i = 1, 2.

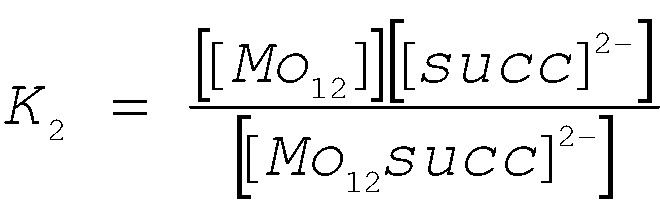
Then, a Van’t Hoff treatment of lnKi (i = 1, 2) versus 1/T (shown in Fig. 4B) allowed to extract the standard enthalpy for the reactions 1 and 2; the standard entropy was then calculated from the Gibb’s function. The calculated thermodynamic parameters for both successive succinate transfers are given in Table 3. The negative values found for enthalpy and entropy for Eqs. 1 and 2 could be tentatively attributed to the formation of the hydration sphere of the produced ionic species, i.e. the uncoordinated succinate ions. Such an interpretation would reflect the main contribution of the polar water molecules. In fact, the origin of the driving force governing the inclusion of the dicarboxylate within the cavity is the entropy increase.
Thermodynamic parameters for the both successive succinate transfers from [Mo12-succ2]4–
| Eqs. | ΔrH° (kJ mol–1) | ΔrS° (J K–1 mol–1) |
| 1 | –29.1 ± 0.2 | –158 ± 1 |
| 2 | –55.7 ± 0.5 | –251 ± 2 |
2.2.6 Variable-temperature 1H NMR in CD3CN
The 1H NMR spectra of Li4[Mo12-succ2]·n H2O is presented in Fig. 5. In CD3CN, the spectra for both compounds, Li4[Mo12-succ2]·n H2O (not shown) and Li2[Mo12-fum]·n H2O (Fig. 5) contain three types of signal related to the three types of protons present in the alkyl chain, in water and in the hydroxo bridges of the ring [14]. For both the compounds, at 303 K, the protons of the alkyl chain give one single sharp resonance at –0.54 and + 3.27 ppm for succinate and fumarate in Mo12-ring, respectively. The deshielded domain exhibits one broad symmetric signal at about +9 ppm which corresponds to the hydroxo groups of the inorganic wheel. As the temperature decreases from 303 to 227 K, the signal of the hydroxo bridges remains single but sharpens drastically from about 50 to 5 Hz. Similarly, the peak of the water at 3.2 ppm exhibits the same type of behavior, revealing a dynamic exchange between protons of the rings and those of the water. Although those results cannot be entirely explained (only one signal for the hydroxo bridges is observed, instead of the two expected), they argues for a single host–guest conformation, likely due to impossibility of the substrate to wheel inside the cavity. In the [Mo12-fum]2– anion, the rigidity of the linear chain probably cancels any concerted hopping of both the carboxylato groups, while in the [Mo12-succ2]4–, the presence of two guests, closely arranged in the cavity prevents any conformational change of both the succinate ions.

Variable-temperature 1H NMR of [Mo12S12O12(OH)12(OH2)4 (C4H2O4)]2– (2) as Li salt in CD3CN. The peaks marked with an asterisk (*) correspond to the solvent (CHD2CN).
3 Discussion
These results exemplify the potentialities of such a host–guest system and give two additional compounds in the series of the dicarboxylate templated rings. Relationships between the size of the inner linear chain and the nuclearity of the ring are evidenced and merit to be discussing herein. C2, C4, C5, C7 and C9 linear saturated chain of dicarboxylate ions have provide rings with variable nuclearity from eight to 16 with M=Mo and W. In this system, the templated rings are of two types, i) the mono-templated rings as [Mo10-glu]2–, [Mo12-pim]2– [14], [Mo14-aze]2– [19] and the bis-templated as [Mo12-succ2]4– and [W16-glu2]4– [13]. For the Mo series, the dependence of the nuclearity of a ring (y) with respect of the number of carbon atoms (x) in the alkyl chain is graphically shown in Fig. 6. The odd series with the C5, C7 and C9 chains gives exclusively the mono-templated Mo10, Mo12 and Mo14 rings, respectively, and provides a linear correlation (a) (y = x + 5) presented in Fig. 6. With the succinate ion, a C4 even alkyl chain, the Mo8 is obviously too small for containing the C4 chain, while the C4 chain is expected also to be too small for a Mo10 ring, which perfectly matches with a C5 chain [14]. In this case, the expansion of the ring is observed for giving a bis-templated [Mo12-succ2]4–. This observation could be extended for the prediction of few additional structures. For the previous reasons, the even C6 and C8 alkyl chains cannot give neither Mo10, Mo12 nor Mo14 mono-templated rings. An expansion of the ring is then expected with such dicarboxylate ions for giving bis-templated arrangements. According to the equation (a), the increasing of two carbon in the chain provokes the increase of 2 Mo in the ring. For the bis-templated ring, the two carbon extension of both the C2n chains should be accompanied by the expansion of 4 Mo, giving the (b) curve in Fig. 6. Finally, the bis-templated series could be described by the curve (b) in Fig. 6, giving the postulated Mo16 and Mo20 rings with C6 and C8 linear dicarboxylate ions, respectively. Recently, some preliminary results we obtained with C6 ion (elemental analysis) seem to confirm the formation of the bis-templated Mo16 ring. Studies are currently in hand to provide unambiguous characterizations for the Mo16 and Mo20 bis-templated rings.
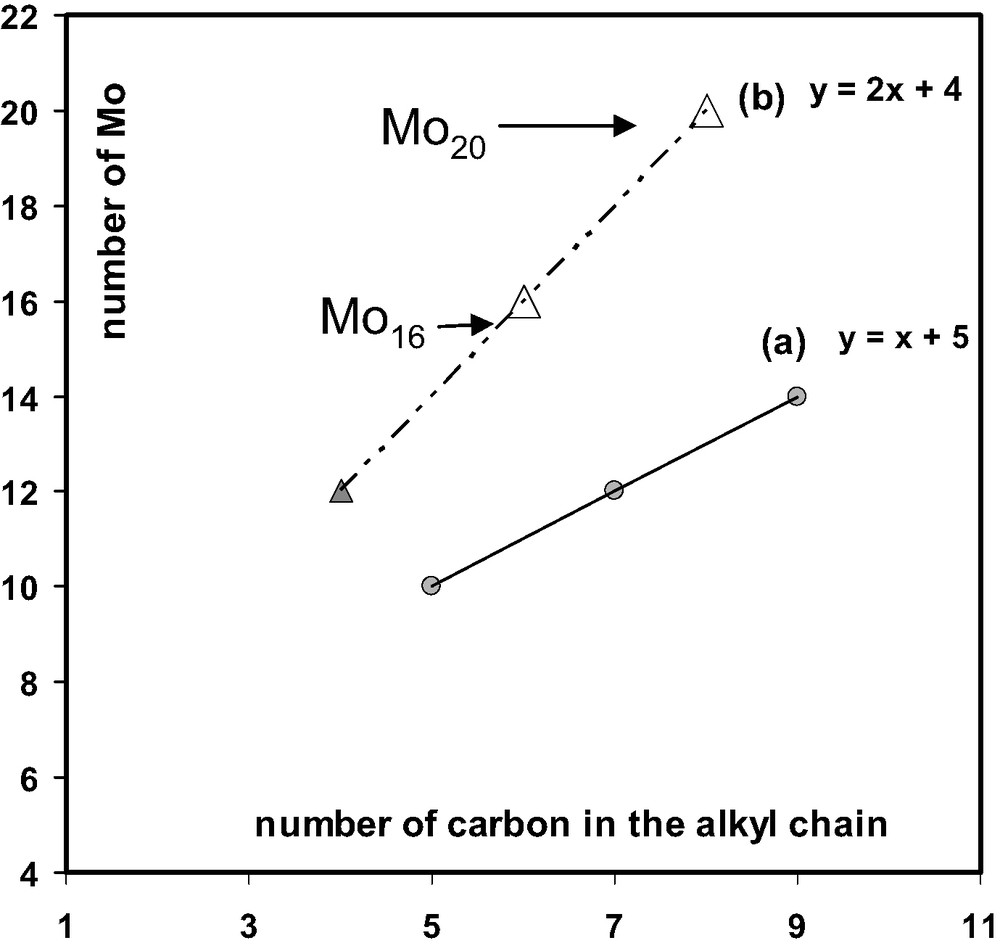
Plots of the nuclearity of the Mo2n ring versus the length of the alkyl chain: (a) odd series giving the mono-templated rings; (b) even series leading to the bis-templated rings, revealing two predicted compounds, the Mo16 and Mo20 rings.
4 Concluding remarks
This work presents two novel templated-Mo12-rings with fumarate and succinate ions. Beside the structural characterizations, 1H NMR studies in solution gave some fundamental insights about thermodynamic of the template exchange within the rings. Those results should be considered for the synthesis of more sophisticated hybrid molecular materials. Finally, some reasonable predictions can be made in the field of our own knowledges.
5 Supporting information available
Further details of the crystallographic data of 1 and 2 can be obtained from the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 IEZ, UK (fax: +44-1223-33-6033 or e-mail: deposit@cdc.cam.ac.uk) as supplementary publication on quoting the depository number, No. CCDC-244995 for 1 and No. CCDC-244996 for 2.
Acknowledgements
Ludovic Lecroq, M.Sci. student, is acknowledged for his contribution to the experimental work.