1 Introduction
Heteropolytungstates of the Wells–Dawson structural class, including their transition-metal-substituted derivatives, are a large and rapidly growing class of polyoxometalates (POMs) with applications in catalysis (acid and oxidation), medicine, and molecular magnetism [1–5]. Many of the fundamental properties of POMs that impact their applications, including elemental composition, solubility, redox potential(s), charge density, size, and shape, can be systematically altered to a considerable degree. An important subset of this entails the development of rational methods for the selective substitution of the d0 metal atoms that form the framework of POM structures. One example of site selective metal substitution was reported by Contant and co-workers on the Mo(VI)- and V(V)-substituted derivatives of the Wells–Dawson complex α-[P2W18O62]6– [6–8]. These investigators were able to control metal substitution through the stereospecific preparation of the defect (i.e. lacunary) species (α1-[P2W17O61]10– and α2-[P2W17O61]10–, for example).
More recently, we and others have reported metal substitution reactions of the divacant lacunary sandwich-type species, αααα-[(NaOH2)2(FeIII)2(X2W15O56)2]16–, where X = P(V) or As(V) [9–11]. These compounds react with Ni(II), Mn(II), or Co(II) under mild conditions and in the presence of an excess of the transition metal to form the mixed-metal complexes, αββα-[(MIIOH2)2(FeIII)2(X2W15O56)2]14–, where M = Ni, Mn, or Co and X = P(V) or As(V). Upon incorporation of two equivalents of Ni(II), Mn(II), or Co(II) there is a 60° rotation of each central α-junction to a β-junction. This rotation (i.e. Baker–Figgis isomerization) is attributed to the fact that a [(MIIOH2)2(FeIII)2(X2W15O56)2]14– structure having two central α-junctions would have steric repulsion between the external MII-OH2 units and the bridging oxygen atoms of the neighboring belt W(VI) atoms [9,12].
We now report the reaction of Zn(II) with the diferric Wells–Dawson sandwich-type complex, αααα-[(NaOH2)2(FeIII)2(X2W15O56)2]16– (X = P (Fe2P4) or As (Fe2As4)), to give αββα-[(ZnIIOH2)2(FeIII)2(X2W15O56)2]14– (X = P (Zn2Fe2P4) or As (Zn2Fe2As4)) (Fig. 1). These complexes are characterized primarily by infrared spectroscopy, elemental and thermogravimetric analyses, X-ray crystallography, differential scanning calorimetry (DSC), and cyclic voltammetry. Electrocatalytic studies confirm that Zn2Fe2As4 is a superior catalyst to Zn2Fe2P4 for the reduction of H2O2, the reverse being true for the reduction of O2.
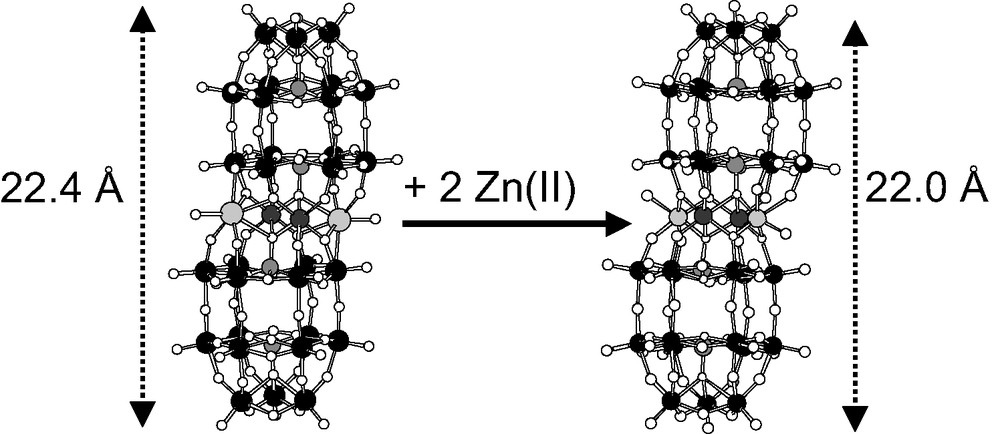
Reaction of Fe2X4 with Zn(II) yields Zn2Fe2X4. The polyoxometalates (POMs) are shown in ball-and-stick notation.
2 Experimental section
2.1 General methods and materials
αααα-[(NaOH2)2(FeIII)2(P2W15O56)2]16– and αααα-[(NaOH2)2(FeIII)2(As2W15O56)2]16– were prepared by the literature methods, and their purity was checked by infrared spectroscopy and cyclic voltammetry [10,13]. As, Fe, Na, P, W, and Zn analyses were performed by Kanti Labs (Mississauga, Canada). Infrared spectra (2% sample in KBr) were recorded on a Nicolet 510 FTIR instrument. Average magnetic susceptibilities were measured on a Johnson–Matthey Model MSB-1 magnetic susceptibility balance as powders at 24 °C; the balance was calibrated using Hg[Co(SCN)4] as a standard. Pascal’s constants were used to obtain the final diamagnetic corrections. Differential scanning calorimetric and thermogravimetric data were collected on ISI DSC 550 and TGA 1000 instruments, respectively. 31P NMR spectra were recorded on a Varian INOVA 400 MHz spectrometer using 85% H3PO4 as an external reference. 183W NMR spectra were recorded on a Varian UNITY 400 MHz spectrometer using 0.2 M Na2WO4·2 H2O (WO42– (aq), δ = 0 ppm) as an external reference. For electrochemical studies, solutions were de-aerated with Ar for 30 min prior to measurements and kept under positive pressure at all times. Pure NO (N20 grade) was purchased from Air Liquide (France). The concentration of the POM in solution was 2 × 10–4 M, unless otherwise stated. The source, mounting, and polishing of the glassy carbon electrodes (GC, Tokai, Japan, 3 mm diameter) have been described in previous work [14]. The counter electrode was a platinum gauze of large surface area. The electrochemical apparatus was a EG and G 273A under computer control (M270 software). All experiments were performed at ambient temperature, and potentials are quoted against a saturated calomel electrode (SCE). Controlled potential coulometry determinations were performed as described for the cyclic voltammetry experiments except that the working electrode used was a 4 cm2 surface area glassy carbon plate. In addition, after deaeration, Ar was continuously bubbled through the solution to ensure a constant stirring of the mixture.
2.2 Synthesis of Na14[(ZnIIOH2)2(FeIII)2(P2W15O56)2]
A 1-g sample of Fe2P4 was added to a 10-ml solution of ZnCl2 (0.035 g, 0.25 mmol) (or Zn(NO3)2·6 H2O (0.074 g)) dissolved in 1 M NaCl, and the mixture was heated to 65 °C for 30 min. The resulting solution was slowly cooled to room temperature, and a microcrystalline precipitate formed after 1–3 days (0.6 g, yield 61%). The precipitate was re-dissolved in 0.5 M NaCl to give diffraction-quality crystals. IR (2% KBr pellet, 1200–500 cm–1): 1088 (s), 1064 (m, sh), 1017 (w), 950 (m, sh), 942 (s), 923 (w), 894 (w), 826 (m), 764 (w), 598 (w), 576 (w), and 524 (m). Magnetic susceptibility: μeff = 9.71 μB mol–1 at 297 K. 31P NMR (in D2O): δ = –12.3 ppm, Δν1/2 = 100 Hz. Anal Calcd for Na14(ZnIIOH2)2(FeIII)2(P2W15O56)2·48 H2O (for a sample dried in air for 12 h): Fe, 1.23; Na, 3.56; P, 1.37; W, 61.0; Zn, 1.44. Found: Fe, 1.29; Na, 3.68; P, 1.41; W, 61.0; Zn, 1.50. [MW = 9051 g mol–1].
2.3 Synthesis of Na10H4[(ZnIIOH2)2(FeIII)2(As2W15O56)2]
A 1-g sample of Fe2As4 was added to a 10-ml solution of ZnCl2 (0.035 g, 0.25 mmol) dissolved in 1 M NaCl, and the mixture was heated to 65 °C for 30 min. The resulting solution was slowly cooled to room temperature, and a microcrystalline precipitate formed after 1–3 days (0.5 g, yield 52%). The precipitate was re-dissolved in 0.5 M NaCl to give diffraction-quality crystals. IR (2% KBr pellet, 1200–400 cm–1): 1058 (m), 949 (s), 869 (m), 822 (m), 700 (m), 590 (w), 557 (w), 523 (w), and 490 (w). 183W NMR (in D2O): δ = –9.9 ppm, Δν1/2 = 200 Hz (1 W); δ = –46.9 ppm, Δν1/2 = 33 Hz (1 W); δ = –66.2 ppm, Δν1/2 = 20 Hz (2 W); δ = –184.4 ppm, Δν1/2 = 13 Hz (2 W). The resonances of the other tungsten atoms are too close to the paramagnetic Fe(III) centers to give observable peaks. Anal Calcd for Na10H4[(ZnIIOH2)2(FeIII)2(As2W15O56)2]·27H2O: As, 3.42; Fe, 1.27; Na, 2.62; W, 62.9; Zn, 1.49. Found: As, 3.40; Fe, 1.41; Na, 3.01; W, 63.2; Zn, 1.47 [MW = 8761 g mol–1].
2.4 X-ray crystallography
Suitable crystals of Zn2Fe2P4 and Zn2Fe2As4 were coated with Paratone N oil, suspended on a fiber loop, and placed on a Brüker D8 SMART APEX CCD sealed tube diffractometer with graphite monochromated Mo Kα (0.71073 Å) radiation. Data collection, indexing, and initial cell refinements were carried out using SMART software (Version 5.624, Brüker AXS, Inc., Madison, WI). Frame integration and final cell refinements were carried out using SAINT software (Version 6.36A, Brüker AXS, Inc.). Final cell parameters were determined from least-squares refinement on 6903 reflections for Zn2Fe2P4 and 8757 reflections for Zn2Fe2As4. Absorption corrections for each data set were applied using SADABS (Version 2.10, George Sheldrick, University of Göttingen). The structures were determined using Direct Methods and difference Fourier techniques (SHELXTL, V6.12). A complete list of atoms refined anisotropically is provided in the Supporting Information. No H atoms associated with the water molecules of Zn2Fe2P4 and Zn2Fe2As4 were located in the difference Fourier maps. The final R1 scattering factors and anomalous dispersion corrections were taken from International Tables for X-ray Crystallography [15]. Structure solution, refinement, and generation of publication materials were performed using SHELXTL V6.12 software. Additional details are provided in Table 1, and thermal ellipsoid plots are given in the Supporting Information (Figs. S1 and S2).
Crystallographic data and structure refinement for Zn2Fe2P4 and Zn2Fe2As4
| Zn2Fe2P4 | Zn2Fe2As4 | |
| Empirical formula | Fe2Na14O164P4W30Zn2 | Fe2Na10O139As4W30Zn2 |
| Formula weight | 8827.68 | 8511.52 |
| Space group | P | P |
| Unit cell | a = 13.522(3) Å | a = 13.566(3) Å |
| b = 13.799(3) Å | b = 13.807(3) Å | |
| c = 22.830(3) Å | c = 22.767(4) Å | |
| α = 90.443(4)° | α = 90.024(6)° | |
| β = 101.652(4)° | β = 101.783(7)° | |
| γ = 117.428(3)° | γ = 117.935(6)° | |
| Volume | 3678.1(12) Å3 | 3665.3(12) Å3 |
| Z | 1 | 1 |
| Density (calcd) | 3.985 g cm–3 | 3.856 g cm-3 |
| Temperature | 100(2) K | 100(2) K |
| λ | 0.71073 Å | 0.71073 Å |
| μ | 2.4069 cm–1 | 2.4971 cm–1 |
| GOF | 1.067 | 1.000 |
| Final R1a [I > 2σ(I)] | 0.0991 | 0.0836 |
| Final wR2b [I > 2σ(I)] | 0.2416 | 0.2162 |
a R1 = Σ||Fo| – |Fc||/|Fo|.
b wR2 = {Σ[w(Fo2 – Fc2)2]/Σ[w(Fo2)2]}0.5
3 Results and discussion
3.1 Syntheses and physical properties of Zn2Fe2P4 and Zn2Fe2As4
The reaction of the divacant lacunary sandwich-type complexes, αααα-[(NaOH2)2(FeIII)2(X2W15O56)2]16– (X = P (Fe2P4) or As (Fe2As4)), with Zn(II) in 1 M NaCl yields αββα-[(ZnIIOH2)2(FeIII)2(X2W15O56)2]14– (X = P (Zn2Fe2P4) or As (Zn2Fe2As4)) in modest yield (~50–60%) and in high purity. This procedure is somewhat different from that used for αββα-[(MIIOH2)2(FeIII)2(P2W15O56)2]14–, where M = Ni(II) or Mn(II) [9,10]. The key difference is whether the transition metal salt is added to a solution of Fe2X4 (X = P(V) or As(V)) (pH ~ 2) or whether Fe2X4 is added to a solution of the transition metal salt. In the case of Zn(II), the latter procedure is preferred to avoid the formation of small to modest amounts of αββα-[(FeIIIOH2)2(FeIII)2(X2W15O56)2]12– (X = P(V) or As(V)). This may be attributable to pH effects since the pH of the ZnCl2 solution is 4–4.5 (versus 2 for a solution of Fe2X4). It may also be due in part to the general unreactive nature of d10 Zn(II) as 31P NMR studies suggest that the reaction of Fe2P4 with Zn(II) is slower than those observed for Fe2P4 with Ni(II) or Mn(II).
The IR spectra of Zn2Fe2P4 and Zn2Fe2As4 are compared with their divacant lacunary precursors Fe2P4 and Fe2As4 in Fig. 2. The W = O (W–O) stretching and W–O–W bending vibrations are quite similar in all four compounds suggesting that the incorporation of Zn(II) into Fe2X4 (X = P(V) or As(V)) has mimimally perturbed the framework of the α-[X2W15O56]12– moieties. The splitting of the ν3 vibrational mode of the central [PO4]3– unit observed in Fe2P4 is removed upon incorporation of the two Zn(II) cations. This suggests that the Td symmetry of the [PO4]3– unit is nearly restored upon binding Zn(II) [16–18].
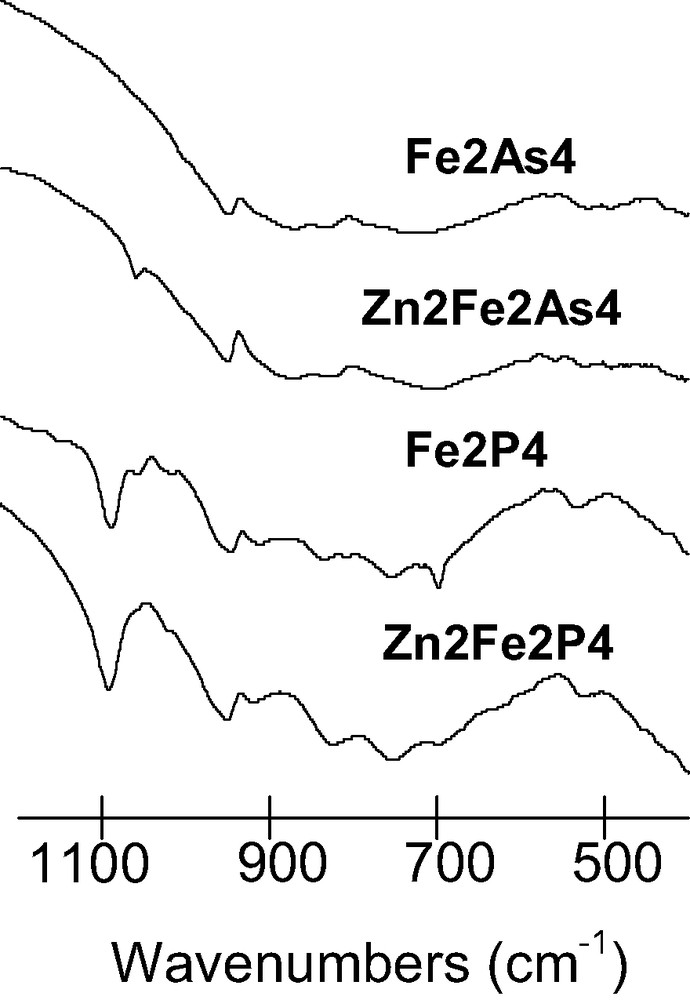
Infrared spectra of Fe2As4, Zn2Fe2As4, Fe2P4, and Zn2Fe2P4.
Differential scanning calorimetry data of Zn2Fe2X4 and Fe2X4 are consistent with their molecular formulas, analytical data, and their crystal structures (Fig. 3). A series of endotherms are observed between 25 and 250 °C in all four complexes that are attributable to the loss of solvent water molecules and coordinated aqua ligands. These data are corroborated by thermogravimetric analyses over the same temperature range. In addition, a series of exotherms are observed between 450 and 600 °C which may be attributable to the crystallization of tungsten oxide and/or the decomposition of the tungsten–oxygen framework of the α-[X2W15O56]12– moieties. The infrared spectra of the complexes show significant changes in the W = O (W–O) stretching and W–O–W bending modes upon heating the samples between 450 and 600 °C. The P analogues (Fe2P4 and Zn2Fe2P4) show a single exothermic process (Fig. 3A) whereas the As analogues (Fe2As4 and Zn2Fe2As4) have a series of stepwise exotherms (Fig. 3B). This is most likely due to heteroatom effects (P versus As) since structurally the two pairs of complexes (Fe2X4 and Zn2Fe2X4) are virtually identical. Interestingly, the divacant lacunary complex, Fe2P4, is more stable in the solid state than the product Zn2Fe2P4. This is surprising given the fact that Fe2P4 has a higher negative charge than Zn2Fe2P4 (16– vs. 14–, respectively). However, Fe2P4 contains four α-junctions whereas Zn2Fe2P4 contains two α-junctions and two β-junctions. Given the fact that α-junctions were previously determined to be 4 kcal mol–1 more stable than β-junctions for dehydrated, solid samples of (NH4)6[P2W18O62], these results suggest that the type of junctions present in the sandwich-type structures plays a key role in their stability [19]. In addition, the steric repulsion present between the external MII-OH2 units and the bridging oxygen atoms of the neighboring belt W(VI) atoms (most prominent in α-junctions) would no longer be a factor since the aqua ligands are lost at temperatures higher than 250 °C.
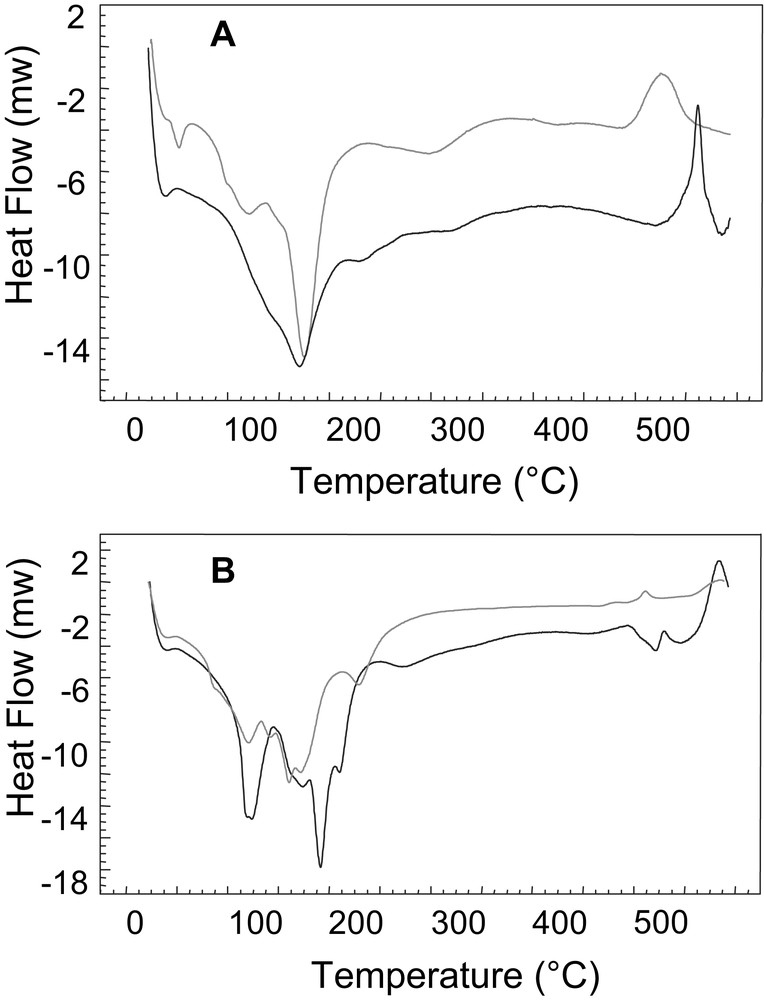
(A) Differential scanning calorimetry data for Zn2Fe2P4 (gray line) and Fe2P4 (black line). (B) Differential scanning calorimetry data for Zn2Fe2As4 (gray line) and Fe2As4 (black line).
3.2 Crystallographic studies
The X-ray crystal structure of Zn2Fe2P4 reveals that a tetranuclear iron-zinc core, [FeIII2ZnII2O14(H2O)2]18–, is sandwiched between two trivacant Wells–Dawson α-[P2W15O56]12– moieties. In addition, two Zn(II) atoms have replaced the two Na+ atoms in the central unit of Fe2P4 with simultaneous 60° rotation of the structure to the conventional inter-POM-unit connectivity known as β (i.e. β-junctions). Each Zn(II) atom is ligated by three oxygen atoms from one of the two α-[P2W15O56]12– units, two oxygen atoms from the other α-[P2W15O56]12–, and a terminal aqua ligand. Bond valence sum calculations from the X-ray crystal structure of Zn2Fe2P4 give an average oxidation state of 2.92 ± 0.03 and 2.09 ± 0.03 for the two Fe and the two Zn atoms, respectively [20].
The X-ray crystal structure of Zn2Fe2As4 reveals that it is nearly isostructural to Zn2Fe2P4, except that the As–O bond lengths are longer than those observed for the analogous P–O bond lengths in Zn2Fe2P4. Overall, this has not caused significant perturbation of the tungsten-oxygen framework of the α-[X2W15O56]12– (X = P(V) or As(V)) moieties or the central [FeIII2ZnII2O14(H2O)2]18– unit as revealed by a comparison of selected average bond lengths and angles summarized in Table 2. Bond valence sum calculations from the X-ray crystal structure of Zn2Fe2As4 give an average oxidation state of 3.03 ± 0.02 and 2.03 ± 0.02 for the two Fe and the two Zn atoms, respectively [20]. There is rotational disorder in both of the polar W3O13 caps of Zn2Fe2As4 with two different conformations present (αα and ββ). This type of cap disorder is very common in Wells–Dawson sandwich-type structures. Interestingly, cap disorder is also observed in solution by 183W NMR. These studies show that two pairs of peaks (δ = –9.9 ppm (1 W) and –184.4 ppm (2 W) for one cap configuration and δ = –46.9 ppm (1 W) and –66.1 ppm (2 W) for the other configuration. The usual disorder of cations and solvent molecules makes it impossible to account for their total numbers in Zn2Fe2As4 by X-ray crystallography alone. As a result, elemental and thermogravimetric analyses were used to determine the total number of Na+ cations and water molecules, respectively.
Selected average bond lengths (Å) and bond angles (°) for Zn2Fe2X4a
| Zn2Fe2P4 | Zn2Fe2As4 | |
| W–Ot | 1.718(7) | 1.719(12) |
| W–Ob | 1.905(6) | 1.910(12) |
| X–O | 1.547(6) | 1.683(12) |
| Zn–OH2 | 2.014(7) | 2.030(15) |
| Zn1···Fe1 | 3.240(7) | 3.243(12) |
| Ot–W–Ob (cap) | 102.1(3) | 102.2(6) |
| Ot–W–Ob (belt) | 99.4(3) | 99.4(5) |
a Ot: terminal oxygen; Ob: doubly bridging oxygen; X: P or As.
3.3 Electrochemical studies
Among the mixed-metal complexes characterized to date, αββα-[(MIIOH2)2(FeIII)2(X2W15O56)2]14–, where M = Ni, Mn, or Co and X = P(V) or As(V) reported recently by us [9,10] and by others [11], we have shown that a number of interesting features are displayed by the Mn(II)-containing complexes [10,21]. First, the complexes Mn2Fe2As4 and Mn2Fe2P4 display remarkable stability from pH 0 to 7 in their oxidized forms. Second, electrochemical data indicate that the Fe(II) state within the complexes is stabilized by the presence of the two Mn(II) centers, in contrast with the instability observed for Mn2Fe2P4 synthesized by a different method [11]. Third, the divacant parent complexes Fe2As4 and Fe2P4 were shown to be quite unstable upon electrolysis at pH < 4, in the absence of Mn(II) [22]. Finally, striking cooperativity effects were observed between the Fe and Mn centers in the electrocatalytic reduction of nitrite and nitric oxide [21]. Therefore, we wondered whether substitution of two Zn(II) into Fe2X4 (X = P or As) might impart similar properties to the resulting complexes analogous to those observed in Mn2Fe2As4 and Mn2Fe2P4.
The stability of Zn2Fe2As4 and Zn2Fe2P4 as a function of pH and time was evaluated by monitoring their UV–vis spectra between 200 and 600 nm over at least 24 h. The As-derivative, Zn2Fe2As4, was found to be stable from pH 0 to 7, and Zn2Fe2P4, at the very least, was stable from pH 0 to 6. Only a small amount of decomposition (ca. 12%) was observed after 24 h for Zn2Fe2P4 at pH 7. The stability of the complexes was also evaluated by cyclic voltammetry and the results were found to be complementary to those obtained from the electronic spectral data.
Fig. 4A compares the cyclic voltammograms of Zn2Fe2As4 and Zn2Fe2P4 in a pH 3 medium (0.5 M Na2SO4 + H2SO4). Table 3 gathers the reduction peak potentials measured from Fig. 4A. The expected overall shift of the voltammogram in the positive potential direction upon the replacement of the P heteroatoms with As in the POMs is once again observed in these studies as well. The shifts are less pronounced for the Fe-waves than they are for the W-waves. The complexes, Mn2Fe2As4 and Mn2Fe2P4, display analogous cyclic voltammograms, published previously [10,21] and with the corresponding peak potential values spanning the same range as those obtained here. In short, in all of the present M2Fe2X4 complexes (M = Zn(II) or Mn(II); X = P or As), the Fe- and W-wave systems were found to be well-separated from each other, facilitating the sequential study of each of the two groups of redox centers. All of the complexes display the classical behaviors usually encountered for the W-centers in the sandwich-type Wells–Dawson complexes. Therefore, in this manuscript attention will be focused primarily on the Fe-waves that are observed at more positive potentials than the W centers. There are two specific features about the Fe-centers which we will focus on most closely: the stability of the Fe(II) state within the complexes and their electrocatalytic properties. Controlled potential coulometry at potentials just negative of the peak potential of the second Fe-wave or the combined single Fe-wave (vide infra) consumed 1.95 ± 0.07 electrons per molecule thereby establishing the oxidation state of the two Fe(III) centers in the freshly prepared complexes.
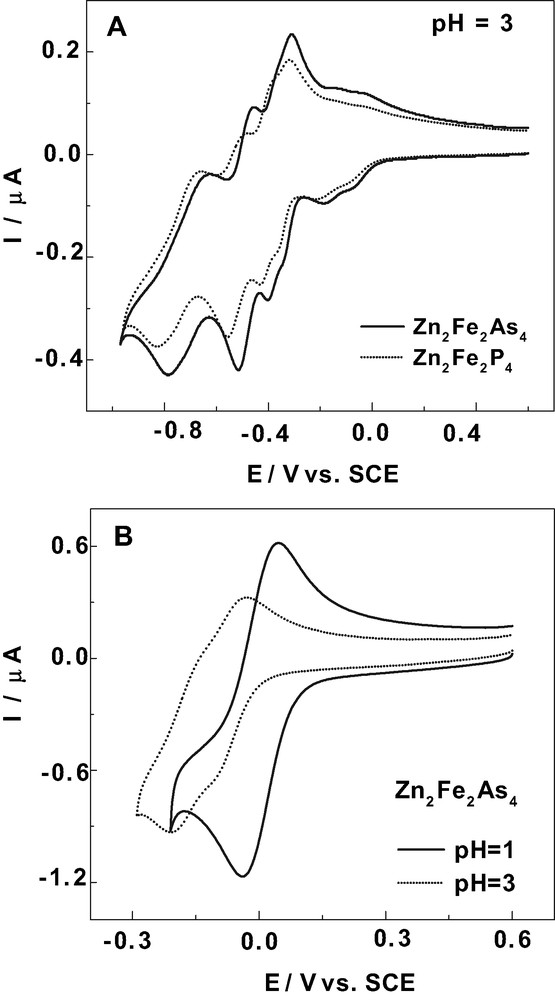
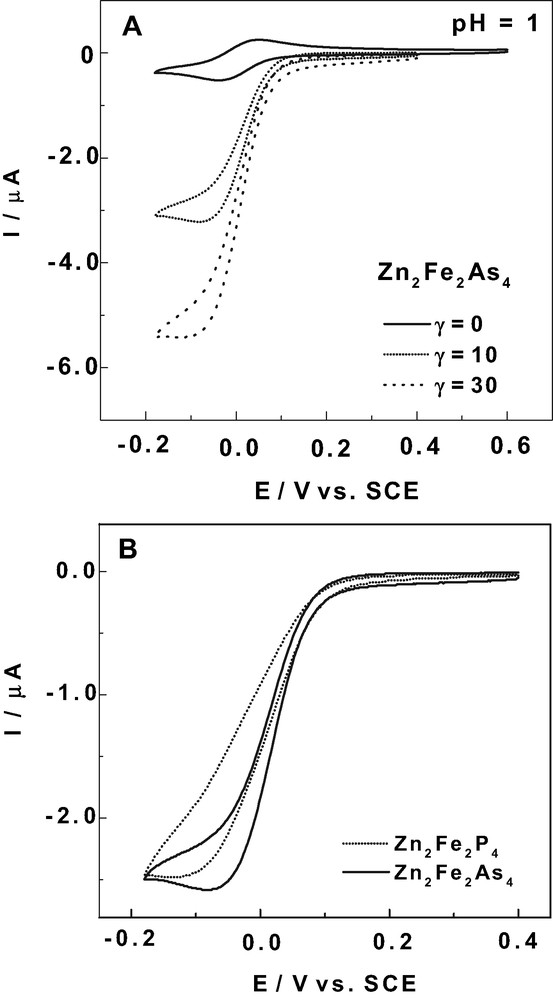
(A) Cyclic voltammograms of 2 × 10–4 M solutions of Zn2Fe2As4 and Zn2Fe2P4 in a 0.5 M Na2SO4 + H2SO4 (pH 3) medium. (B) Cyclic voltammograms of 2 × 10–4 M solutions of Zn2Fe2As4 restricted to the Fe(III)-based waves in pH 1 and pH 3 media (0.5 M Na2SO4 + H2SO4). The scan rate was 10 mV s–1, the working electrode was glassy carbon, and the reference electrode was SCE.
Reduction peak potentials at pH 3 (0.5 M Na2SO4 + H2SO4) for Zn2Fe2X4 (X = P or As)a
| Complex | – Epc/mV | ||
| Fe1 | Fe2 | W-waves | |
| Zn2Fe2As4 | 86 | 186 | 340; 400; 512; 782 |
| Zn2Fe2P4 | 96 | 198 | 370; 423; 536; 828 |
a The values were measured from the cyclic voltammograms run at a scan rate of 10 mV s–1. The reference electrode was SCE. The Fe centers are numbered arbitrarily 1 and 2 for clarity from the less negative to the more negative potential.
A representative example of the behaviors of the Fe-waves as a function of pH is shown for Zn2Fe2As4 in Fig. 4B. The formerly split Fe-waves at pH 3 now merge at pH 1, indicating a clear influence of the acidity of the medium. Analogous observations were made with Zn2Fe2P4, but a slightly composite wave is obtained at pH 1, with the coalescence being somewhat incomplete. The results suggest that there is a pronounced influence from the central heteroatom, P or As. A comparison among all of the present M2Fe2X4 complexes (M = Zn(II) or Mn(II); X = P or As), leads to the general conclusion that slight differences in their voltammetric behaviors are also a result of the different cations present within the mixed-metal complexes (i.e. Zn(II) or Mn(II)). These cations can modulate the acid–base properties of the complexes and correlatively their redox behaviors.
3.4 Electrocatalytic studies (evolution and progress)
In the following studies, we are interested in evaluating the electrocatalytic processes related to the redox and/or chemical properties of the POMs. The electrocatalytic behaviors of modified surfaces in which the basic properties of the POMs themselves cannot be distinguished are not considered [23]. The NOx family is taken as an example. Electrocatalysis of the reduction of HNO2 and NO was demonstrated in the late 1980s independently by Toth and Anson [24] and by our group [25,26]. Since then, this reaction has been selected and is being used worldwide as a classical test of the electrocatalytic properties of the POMs. Consideration of [Fe(H2O)SiW11O39]6– (SiW11Fe for short), one of the electrocatalysts proposed by Toth and Anson [24], is particularly enlightening: a Fe-nitrosyl complex is formed upon reduction of the Fe center and the actual catalytic process occurs at more negative potentials where the necessary number of charges is accumulated and delivered by the W-framework. However, a loss in selectivity and a low yield ensue [24] from electrode derivatization [23]. Recently, to avoid the shortcomings encountered with SiW11Fe, we and others have proposed several alternative POMs [14,25–28]. Among all of these potentially acceptable candidates, we have obtained N2O with a quantitative yield of 100% with molybdic and molybdo-tungstic POMs [27]. A supramolecular compound like [P8W48O184]40–, with only W centers, can also be used to reduce NO efficiently without derivatization of the electrode surface [29]. Finally, sandwich-type complexes have the advantage of accumulating metal centers known for their catalytic behaviors in the reduction of NOx, and they permit a substantial positive shift of the potential because these metal centers are easier to reduce than the W centers of the polytungstic framework [21,30–32].
In this context, electrocatalytic reduction of HNO2 and NO by the present M2Fe2X4 (M = Mn(II) or Zn(II); X = P or As) complexes constitutes a further step in the search for efficient electrocatalysts. Fig. 5A shows the current enhancements accompanying the addition of increasing amounts of NaNO2 to a solution of Zn2Fe2As4 at pH 11. This pH 1 value was selected because it ensures a fairly complete merging of the two Fe-waves in this complex. The increase in the current intensity is readily observed at the reduction potential of the Fe(III) centers for modest increases in the γ values (γ is the excess parameter defined as γ = C° (NOx)/C° (POM)). In this potential domain, no direct reduction is observed for the NO or HNO2 present in solution [27,33]. Furthermore, it is worth noting that all of the electrocatalysts show high efficiency even though modest γ values were used. Similar observations were made with pure NO as those obtained for nitrite in reasonably acidic solutions. Therefore, the corresponding results will not be described separately [21,27,33].
Fig. 5B compares the electrocatalytic HNO2 reduction waves for Zn2Fe2X4 (X = P or As) with an excess parameter γ = 10. The pattern for the As-derivative was shifted to a more positive potential domain compared to that for the P-derivative, even though the two final plateau currents are the same. Correlatively, for a selected potential value, the electrocatalytic activity of Zn2Fe2As4 remains higher than that of its P-analogue up to the plateau current of the latter. The catalytic efficiency (CAT) is defined as follows: CAT = 100 × [I (POM + NO or HNO2) – Id(POM)]/Id(POM), where I(POM + NO or HNO2) is the current for the reduction of the POM in the presence of NO or HNO2 and Id (POM) is the corresponding diffusion current for the POM alone. This parameter must be considered with care. A rigorous comparison of the CAT values between two or more electrocatalysts demands that the same potential be selected to measure the currents (and eventually, the same catalytic current if an improvement in the potential is to be evaluated). Here, for γ = 10 and at a potential of –50 mV versus SCE, CAT values are 696% for Zn2Fe2P4 and 1002% for Zn2Fe2As4. This result parallels that obtained for Mn2Fe2X4 (X = P or As), where the CAT value was approximately 25% higher for the As-derivative than for the P-analogue [21]. Although the attention here is focused mainly on the behaviors of these mixed-metal sandwich-type compounds, comparable catalytic efficiencies were also obtained under similar conditions for the lacunary precursors, Fe2As4 and Fe2P4 [21]. However, in contrast to the mixed-metal complexes, the stability of the Fe(II) states within Fe2As4 and Fe2P4 in acidic media is very low[22], and the data suggests that the mixed-metal complexes are better choices for long-term assays.
Experiments with NaNO2 at pH 5 where HNO2 and NO do not form, show that the catalytic process was relegated to the reduction potential of the first W-wave of the present mixed-metal complexes. In contrast, the catalysis starts directly in the iron-reduction potential domain when pure NO was used at pH 5. These observations are similar to those previously made with Mn2Fe2X4 (X = P or As) [21]. However, the CAT value measured at –400 mV versus SCE in the iron-reduction potential domain is about six times larger for Mn2Fe2As4 than for Zn2Fe2As4.
In summary, there are three features in the preceding results that are favorable for the use of these POMs in the electrocatalytic reduction of HNO2 and NO. First, these POMs are stable from pH 0 to at least 6, a feature not shared by the otherwise efficient molybdic or molybdo-tungstic POMs. Second, these complexes show the enhanced stability of the Fe(II) state within the mixed-metal complexes relative to those observed in their lacunary precursors, Fe2As4 and Fe2P4. Finally, the third feature exhibited by these complexes which is favorable for electrocatalytic processes is the ability of the Fe-based redox waves to trigger the catalytic processes without any direct interference from the W(VI) centers.
A pH 5 medium was selected for the studies of the electrocatalytic reductions of O2 and H2O2. The efficiency of the catalysis is again evaluated by an excess parameter γ, which is defined as C°(O2)/C°(POM) for dioxygen and C°(H2O2)/C°(POM) for hydrogen peroxide (C = concentration of the relevant species indicated in parentheses). The catalytic efficiency (CAT) is defined as follows: CAT = 100 × [I(POM + O2 or H2O2) – Id(POM)]/Id (POM), where I (POM + O2 or H2O2) is the current for the reduction of the POM in the presence of O2 or H2O2 and Id (POM) is the corresponding diffusion current for the POM alone. Selected values of catalytic efficiencies (CAT) for the reduction of dioxygen and hydrogen peroxide are included in Table 4. CAT values were measured at –420 mV versus SCE, in the potential domain of the Fe(III)-reduction waves.
Selected values of catalytic efficiencies measured at E = –420 mV vs. SCE in a pH 5 medium (1 M CH3COOLi + CH3COOH) for O2 and H2O2 reductions with various electrocatalystsa
| Complex | CAT | |
| O2 | H2O2 | |
| Zn2Fe2As4 | 680 | 334 |
| Zn2Fe2P4 | 817 | 222 |
| Mn2Fe2As4 | 944 | 464 |
| Mn2Fe2P4 | 1055 | 380 |
a The scan rate was 2 mV s–1. The catalytic efficiency (CAT) is defined as follows: CAT = 100 × [I(POM + O2 or H2O2) – Id(POM)]/Id(POM), where I(POM + O2 or H2O2) is the current for the reduction of the POM in the presence of O2 or H2O2 and Id(POM) is the corresponding diffusion current for the POM alone.
Corresponding CAT values for Mn2Fe2X4 (X = P or As) were also measured and included for comparison. Under the same experimental conditions, the phosphorus-containing complexes, M2Fe2P4 (M = Zn(II) or Mn(II)), are more efficient than their arsenic analogues, M2Fe2As4 (M = Zn(II) or Mn(II)), for dioxygen reduction, while the opposite is true for hydrogen peroxide reduction. This observation underscores the difficulty for anticipating which electrocatalyst would be more efficient when an inner sphere pathway is operative. The electrocatalytic processes observed here remain efficient in other media, and they establish the utility of the Fe(III) centers within these mixed-metal sandwich complexes even in media of low proton availability. There is still ample room for optimization of the selected media for efficient electrocatalytic processes within these compounds.
Examination of the two electrocatalytic processes discussed here may also shed some light on possible reaction pathways. Dioxygen does not coordinate to Fe(III) but it does coordinate to Fe(II). Then, the presence of another reduced Fe center could drive the catalytic process to completion by carrying out immediate further reduction of the Fe–O2 adduct. Also noteworthy is that the synthesis of M2Fe2X4 (M = Zn(II) or Mn(II); X = P or As) from Fe2X4 (X = P or As) diminishes the overall negative charge of the POM, rendering its reduction more facile, and ultimately, saving energy during the electrocatalytic process.
4 Conclusions
The previously unknown mixed-metal sandwich-type Wells–Dawson complexes, αββα-[(ZnIIOH2)2(FeIII)2(P2W15O56)2]14– (Zn2Fe2P4) and αββα-[(ZnIIOH2)2(FeIII)2(As2W15O56)2]14– (Zn2Fe2As4), were synthesized and characterized by X-ray crystallography, IR and NMR spectroscopy, elemental and thermogravimetric analyses, differential scanning calorimetry and cyclic voltammetry. In their oxidized forms, they are stable from pH 0 to 6 for Zn2Fe2P4 and from pH 0 to 7 for Zn2Fe2As4. Their Fe-waves are pH-dependent and are observed at more positive potentials than the corresponding ones in their lacunary precursors Fe2X4 (X = P or As). Also unlike Fe2X4, the Fe(II) state is remarkably stable within the mixed-metal complexes. Furthermore, the electrocatalytic reduction of O2, NO and HNO2 occurs readily in the potential domain of the Fe-waves, thus indicating there is multiple electron exchange within this system. The voltammetric behavior of Zn2Fe2As4 and Zn2Fe2P4 are similar to those observed for Mn2Fe2As4 and Mn2Fe2P4. Collectively, these observations suggest that the filling of the vacancies in Fe2X4 (X = P or As) is the main parameter governing the properties of the present mixed-metal sandwich-type complexes, with the identity of the metal (i.e. Zn(II) or Mn(II)) modulating their acid–base and catalytic properties.
Acknowledgments
This work was supported in part by the University Paris-11 and the CNRS (UMR 8000 and 8648), and in part by the NSF (Grant CHE-0236686).
Supplementary material
The structure determination data (in cif form) was sent to Fachinformationszentrum Karlsruhe, Abt. PROKA, 76344 Eggenstein-Leopoldshafen, Germany and may be obtained on quoting depository numbers 414160 (Zn2Fe2P4) and 414159 (Zn2Fe2As4). Thermal ellipsoid plots for Zn2Fe2P4 and Zn2Fe2As4 (in pdf form) may be obtained from the journal office or by contacting the author.
1 At pH 1, the actual active species should be HNO2 and/or NO. In fact, the following sequence is known: HNO2 → H+ + NO2–, pKa = 3.3 at 18 °C and HNO2 disproportionates in fairly acidic solution: 3 HNO2 → HNO3 + 2 NO + H2O. The rate of this reaction is known to be low.