

1 Introduction
There has already been a great deal of attention devoted to the preparation of molecular hybrids based on transition-metal derivatives and tetracyanoethylene (TCNE) or 7,7,8,8-tetracyano-p-quinodimethane (TCNQ) [1,2]. The ability of these polynitrile entities to act as bridging ligands through two or more of their CN groups associated with their strong electron-acceptor properties should afford polymeric derivatives displaying original structures associated with remarkable electronic properties [1–8].
In this context, reactivity of TCNX derivatives with dinuclear complexes with a high order metal–metal bond is an area of particular interest since such structure may favour strong electronic interactions within the inorganic fragment [9–11]. To date, however, the literature reports but three examples in the special case of quadruply-bonded compounds. The first one concerns the reaction of TCNE with [Mo2L2] (L = N4 macrocyclic ligand): an electronic transfer without coordination affording [Mo2L2]•+(TCNE)•– was observed [12,13]. The other two are relative to dimolybdenum tetracarboxylate derivatives of general formula [Mo2(O2CR)4]. With R = CMe3, reaction with TCNE results in initial coordination of the polynitrile unit affording after hydrolysis the polymeric compound [Mo2(O2CR)3(OC(NH)C(CN)C(CN)2)]∞ (Scheme 1) [14], whereas with R = CF3, reaction of TCNQ conduces to the polymeric compound [Mo2(O2CCF3)4(TCNQ)]∞, which includes a neutral μ4-TCNQ unit [15].

We report here the preparation and characterisation of a new type of polymeric molecular hybrid including a quadruply-bonded metal–metal unit and a TCNE moiety that uses for coordination with metals only two of its CN groups and behaves so as a μ2-ligand.
2 Results and discussion
Reaction of [Mo2(O2CR)4] (R = CF3) (1) in refluxing toluene with one equivalent of TCNE affords a light bright green compound which corresponds to the formula [Mo2(O2CCF3)4(TCNE)]∞·2 C6H5CH3 (2) (Scheme 1). It is noteworthy that no mild hydrolysis of TCNE, previously observed with the molybdenum pivalate 1′ (R = CMe3), was detected, even by using ‘wet’ toluene or by adding the water into the toluene solvent [14]. As a bulk vacuum-dried solid, 2 is stable in dry air, but the crystals contain toluene and hence disintegrate slowly due to solvent loss when left in the open.
2.1 Crystal structure
Compound 2 crystallises in the hexagonal space group R

ORTEP plot (30% probability level) showing the contents of asymmetric unit in the structure of 2.
Selected bond distances (Å) and angles (°) for compound 2
| Mo–O(3) | 2.111(3) | Mo–Moi | 2.1117(8) |
| Mo–O(2) | 2.114(3) | Mo–O(4) | 2.115(3) |
| Mo–O(1) | 2.116(3) | Mo–N(1) | 2.875(4) |
| O(1)–C(1) | 1.263(6) | O(2)–C(3) | 1.270(6) |
| O(3)–C(1)i | 1.264(6) | O(4)–C(3)i | 1.264(5) |
| N(1)–C(5) | 1.134(6) | N(2)–C(6) | 1.128(6) |
| C(1)–O(3)i | 1.264(6) | C(1)–C(2) | 1.530(7) |
| C(3)–O(4)i | 1.264(5) | C(3)–C(4) | 1.523(7) |
| C(5)–C(7) | 1.479(8) | C(6)–C(7) | 1.464(8) |
| C(7)–C(7)ii | 1.304(11) | C(8)–C(9) | 1.359(9) |
| C(8)–C(13) | 1.386(13) | C(8)–C(14) | 1.512(11) |
| C(9)–C(10) | 1.312(10) | C(10)–C(11) | 1.316(16) |
| C(11)–C(12) | 1.307(18) | C(12)–C(13) | 1.388(18) |
| O(3)–Mo–Moi | 92.10(9) | O(3)–Mo–O(2) | 92.05(12) |
| Moi–Mo–O(2) | 91.94(9) | O(3)–Mo–O(4) | 87.55(13) |
| Moi–Mo–O(4) | 91.68(9) | O(2)–Mo–O(4) | 176.37(12) |
| O(3)–Mo–O(1) | 176.25(12) | Moi–Mo–O(1) | 91.64(9) |
| O(2)–Mo–O(1) | 87.66(12) | O(4)–Mo–O(1) | 92.51(12) |
| O(3)–Mo–N(1) | 91.32(13) | Moi–Mo–N(1) | 172.70(9) |
| O(2)–Mo–N(1) | 94.38(12) | O(4)–Mo–N(1) | 82.02(12) |
| O(1)–Mo–N(1) | 84.98(12) | C(1)–O(1)–Mo | 115.3(3) |
| C(3)–O(2)–Mo | 115.6(3) | C(1)i–O(3)–Mo | 115.1(3) |
| C(3)i–O(4)–Mo | 116.0(3) | C(5)–N(1)–Mo | 166.5(4) |
| O(3)i–C(1)–O(1) | 125.8(4) | O(3)i–C(1)–C(2) | 116.9(4) |
| O(1)–C(1)–C(2) | 117.3(4) | O(4)i–C(3)–O(2) | 124.8(4) |
| O(4)i–C(3)–C(4) | 117.5(4) | O(2)–C(3)–C(4) | 117.7(4) |
| N(1)–C(5)–C(7) | 172.2(6) | N(2)–C(6)–C(7) | 172.8(6) |
| C(7)ii–C(7)–C(6) | 119.5(8) | C(7)ii–C(7)–C(5) | 119.7(8) |
| C(6)–C(7)–C(5) | 120.8(5) | C(9)–C(8)–C(13) | 116.9(7) |
| C(9)–C(8)–C(14) | 121.5(9) | C(13)–C(8)–C(14) | 121.6(10) |
| C(10)–C(9)–C(8) | 122.4(7) | C(11)–C(10)–C(9) | 120.3(9) |
| C(10)–C(11)–C(12) | 122.0(10) | C(11)–C(12)–C(13) | 119.6(9) |
| C(8)–C(13)–C(12) | 118.8(8) |
Within the dimolybdenum skeleton, the paddlewheel structure of the starting complex 1, resulting from the presence of four bridging trifluoroacetate groups, is retained. The Mo–Mo bond distance (2.1117(8) Å) is significantly longer than in 1 (2.090(4) Å) [16]; the slight increase, which is of the same order than that observed in the corresponding TCNQ derivative [Mo2(O2CCF3)4(TCNQ)] (2.1126(8) Å) [15], may be attributed to the axial coordination of the ligand [17,18]. All other bond lengths (Mo–O aver. 2.114; C–O aver. 1.265; C–C aver. 1.527 and C–F aver. 1.317 Å) and bond angles within the inorganic fragment are similar to those observed in the parent compound 1 and do not give rise to further comments [16].
The TCNE entity, the charge state of which is discussed below, presents an almost planar structure and acts, as shown on Fig. 2, as a bridging ligand between two Mo2 units through two trans-1,2-cyano groups. This coordination (Mo–N(1) 2.875 (4) Å) allows the formation of infinite chains (interdimer Mo…Mo distance within the chain: 11.700 Å) resulting in the 1-D structure shown in Fig. 2. It is worthy of note that, in this structure, one observes a μ2-coordination mode of the TCNE unit, while in the TCNQ adduct the polynitrile unit exhibits a μ4-coordination [15]. The TCNE coordination is characterised in 2 by the values of C(5)–N(1)–Mo 166.5(4)° and Moi–Mo–N(1) 172.70(9)° angles which are similar to those observed in analogous rhodium polymer (aver. 164.5° and 172.6° respectively) [18,19] and by a Mo–N bond length which is longer than that usually reported for polymeric [Mo2(O2CCF3)4L2] complexes containing other axial nitrogen donor ligands (2.557 Å for L = 4,4′-bpy [20]; 2.627 Å for L = TCNQ [15]). However, it is worth pointing out that a long Mo–N bond length (2.784 Å) has already been mentioned in the compound [Mo2(O2CCH3)4L] with L = (CH2)6N4 [21].

1-D chain structure present in compound 2.
The overall crystal structure of 2 is built on a quite complex arrangement of infinite 1-D Mo2-TCNE zigzag chains separated by toluene molecules, as illustrated by two packing diagrams (Fig. 3). The complexity of this structure is due to a particular property of R

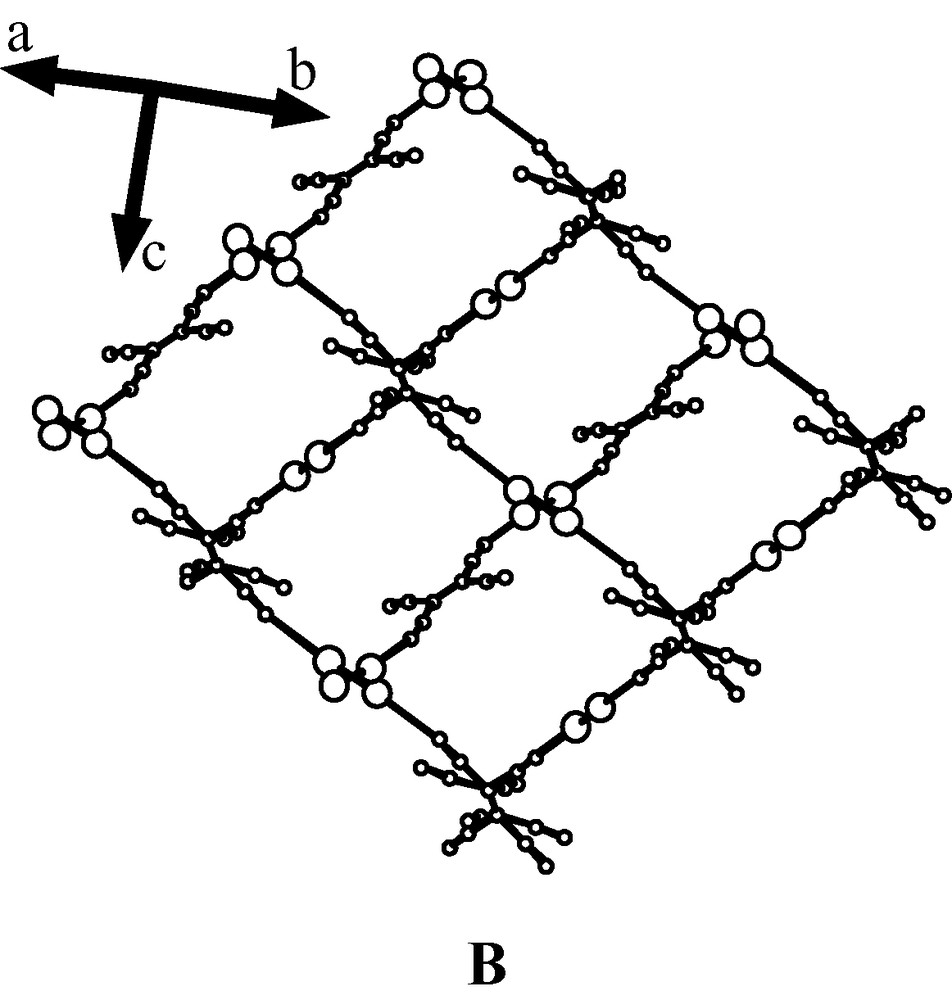
Views of the crossing 1-D chains. For the sake of clarity, O2CCF3 ligands and toluene molecules were removed.

Crossing 1-D chains projected down the c axis showing the screw cavities hosting the toluene molecules. For the sake of clarity, O2CCF3 ligands were removed.
2.2 Charge state of the TCNE ligand
In compound 2, the TCNE unit clearly acts as a neutral ligand as shown by:
- (i) 13C NMR data: the sharpness of the different peaks observed on the well resolved spectrum (see Section 4) indicates a diamagnetic behaviour.
- (ii) Electrochemical data: the first reduction process of TCNE is present on the cyclic voltammogram of 2. This is in total agreement with the relative values of the potentials of the starting materials: the TCNE/TCNE•– potential (E1/2 = –0.20 V vs. the Fc+/Fc reference electrode) is too low to allow an oxidation of 1 (1•+/1, Epox (irr.) = 1.54 V/Fc) [1,22].
- (iii) IR data: the IR spectrum of 2 reveals two ν(CN) bands (2259, 2221 cm–1), the positions of which are very close to those of free TCNE (2260, 2220 cm–1) [23]. It is known that an increase of the negative charge of the TCNE unit results in a lowering of the wave numbers associated with these vibrations (TCNE•–: 2185, 2145; TCNE2–: 2104, 2069 cm–1) [23].
- (iv) Structural data: previous studies show that an increase of the electronic charge on the TCNE unit results in a lengthening of the central C–C bond (neutral TCNE: 1.344 Å [16], TCNE•–: 1.37–1.39 Å, TCNE2–: 1.49 Å) [24]. The bond length observed in 2 (1.304(11) Å) is shorter than that in TCNE, excluding the possibility of a negative charge, even fractional, on the TCNE unit.
2.3 Comparison with other dimolybdenum tetracarboxylate derivatives: why the reaction of TCNE with [Mo2(O2CCF3)4] does not afford the tricyanoacrylamidato derivative?
Formation of a 1/1 adduct in the reaction of TCNE with [Mo2(O2CCF3)4] differs strongly from the hydrolysis reactions observed with [Mo2(O2CCMe3)4] where the final product is the tricyanoacrylamidato (TC3A) compound [Mo2(O2CCMe3)3(OC(NH)C(CN)C(CN)2)] (Scheme 1) [14]. It seems likely that this difference may be ascribed to the high electron withdrawing properties of the CF3 groups as compared to CMe3. This observation leads us to propose, for the hydrolysis reaction, the mechanism which successively involves (Scheme 2):
- (i) an activation of the polynitrile via its weak coordination to the Mo2 core (this step must involve a weak charge transfer from the metal to the organic ligand);
- (ii) a nucleophilic attack of water on the carbocation leading to the formation of the TC3A ligand;
- (iii) a proton transfer from this ligand to the carboxylato group, and then:
- (iv) the decomplexation of the carboxylic acid and its substitution by the TC3A unit.

The difference in behaviour between the two carboxylate derivatives can be rationalised by two concurrent properties of the [Mo2(O2CCF3)4] compound relatively to [Mo2(O2CCMe3)4]. No similar reaction with [Mo2(O2CCF3)4] could arise because: (i) the electron transfer of step (i) is impossible since [Mo2(O2CCF3)4] is much more difficult to oxidise than [Mo2(O2CCMe3)4], and (ii) the proton transfer of step (iii) is impossible since the basicity of the CF3CO2– anion is weaker compared to those of the Me3CCO2– anion.
3 Conclusion
The present finding emphasises the control of the electronic properties and reactivity of the [Mo2(O2CR)4] derivatives by the nature of the carboxylato bridge, leading by reaction with TCNE, either to a donor acceptor [DA] stable material with no charge transfer, or to a TCNE hydrolysed compound after evolution of a [D•+ A•–] intermediate as a transient species.
This represents the two limiting cases of the donor acceptor interaction. Work is in progress aiming to isolate [D•+ A•–] stable extended materials by a rational choice of the linking bridge.
4 Experimental
4.1 Materials and methods
All reactions were performed in Schlenk tubes in a dry dinitrogen atmosphere. Solvents were distilled using standard techniques and were thoroughly deoxygenated before use. IR spectra were obtained with the use of a Nicolet Nexus spectrometer (KBr pellets). 13C NMR spectra were recorded on a Bruker AMX 400 MHz. Elemental analyses were performed by the ‘Service central d’analyses du CNRS’, Vernaison, France. The starting material [Mo2(O2CCF3)4] was prepared as described in the literature [25]. TCNE was purchased from Aldrich and sublimated before use.
4.2 Synthesis of [{Mo2(O2CCF3)4}TCNE] (2)
A stoichiometric mixture of [Mo2(O2CCF3)4] (0.32 g, 0.5 mmol) and TCNE (0.064 g, 0.5 mmol) in toluene (60 ml) was heated at 140 °C in an oil bath for 16 h during which time the solution colour slowly changed from light yellow to pale green. The mixture was cooled to room temperature; the solvent was concentrated to ca. 20 ml, the solution filtered and then left at –20 °C for one night. Bright green crystals were collected. Yield 50% (0.36 g).
Analyses calculated for C14F12Mo2N4O8·1.7C6H5CH3: Calc. C 33.5, H 1.5, N 6.0; found C 33.6, H 1.5, N 5.9; selected IR bands [KBr, cm–1]: 2259w, 2221w (νCN), 1594vs, 1570s (νasCO), 1456m (νsCO), 1190vbr (νCF) cm–1. 13C-NMR (CD3CN): δ 68.5 (s, C=C), 107.5 (s, CN), 108.2 (s, CN), 114.2 (q, 1J(C–F) = 281 Hz, CF3), 167.2 (q, 2J(C–F) = 41.4 Hz, OCO). Cyclic voltammetry (E/V; CH3CN, Bu4NPF6 0.2 mol l–1, ref Fc+/Fc): [MoIII-3.5-MoII(TCNE)]•+/[MoII-4-MoII(TCNE)]: Epox (irre.) = 1.54 V; [MoII-4-MoII(TCNE)]/[MoII-4-MoII (TCNE•)]–: E1/2 = –0.20 V (ΔEP = 60 mV).
4.3 X-ray crystal structure determination for [{Mo2(O2CCF3)4}TCNE]·2 C6H5CH3 (2)
A single crystal of 2 suitable for X-ray studies was mounted on a Nonius Kappa CCD diffractometer. The unit cell determination and data collection were carried out with MoKα radiation (λ = 0.71073 Å) at low temperature (110 K). The measured intensities were reduced with DENZO program [26]. The structure was solved with DIRDIF-99 package [27] and further refined with full–matrix least–squares methods (SHELXL97) based on |F2| [28]. All non-hydrogen atoms were refined with anisotropic thermal parameters. The hydrogen atoms of toluene were included in a riding model and given the isotropic termal parameters of 1.2 times for those beared by sp2 carbon atoms and 1.5 times for those of sp3 methyl carbon. Crystallographic data and final discrepancy factors are gathered in Table 2.
Crystallographic data for compound 2
| Colour | Bright green |
| Shape and size (mm3) | Prism 0.20 × 0.15 × 0.10 |
| Chemical formula | C14H8O4N2F6Mo1 |
| Formula weight | 478.16 |
| Crystal system, space group | Hexagonal, R |
| a (Å) | 20.1697(8) |
| b (Å) | 20.1697(8) |
| c (Å) | 22.0325(12) |
| V (Å3) | 7762.3(6) |
| Z | 18 |
| Dcalc (g cm–3) | 1.841 |
| μ (MoKα) (mm–1) | 0.845 |
| F(000) | 4212 |
| θ range (°) | 1.018; 30.508 |
| hkl ranges | [–27; 19]/[–22; 23]/[–29; 24] |
| Collected reflections, unique reflections, observed reflections, Rint | 9686, 4143, 2713, 0.0519 |
| Reflections, parameters, restraints | 4143, 244, 0 |
| Goodness-of-fit on F2 | 0.953 |
| R(F), Rw(F2)a [I > 2σ(I)] | 0.0514, 0.1247 |
| R(F), Rw(F2)a (all data) | 0.0975, 0.1612 |
| wb/a, b | 0.1000, 0.0000 |
| ρmax, ρmin (e Å–3) | 2.028, –1.303c |
a R(F) = Σ||Fo| – |Fc||/Σ|Fo|, Rw(F2) = [Σw(Fo2 – Fc2)2/Σ[w(Fo2)2]1/2.
b w = 1/[σ2(Fo2) + (aP)2 + bP] with P = (Fo2 + 2Fc2)/3.
c The highest residual peak is found in TCNE ligand 1.0 Å from C7 atom, while the lowest one is 0.88 Å far from Mo atom.
5 Supplementary material
Crystallographic data for the structural analysis have been deposited with the Cambridge Crystallographic Data Centre, CCDC 240184. Copies of this information may be obtained free of charge from the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax +44-1223-336-003; e-mail: deposit@ccdc.cam.ac.uk or www: http://www.ccdc.cam.ac.uk).
Acknowledgments
This work was supported by the CNRS (UMR 6521) and the French ‘Ministère de la Recherche’. MR is acknowledged for doctoral fellowship (BLG). Professor S. Triki (University of Brest, France) is thanked for helpful discussions.