

1 Introduction
Dithiadiazafulvalenes (DTDAF) are interesting π-donor molecules with three oxidation states: neutral, radical cation and dication, where the electron transfer is concerted with molecular movements [1]. Indeed, these compounds are almost planar in the neutral state [2], while in the dicationic form the two thiazole rings of the donor core are twisted around the central C–C bond [1,3]. Therefore, upon oxidation of DTDAF (1), a pronounced rotation of the two thiazole rings around the central C–C bond is observed inducing reduced electronic interactions in the oxidized species [1,3]. Accordingly, a small potential difference (ΔE = E2 – E1) between the two oxidation potentials is observed (E1: DTDAF/DTDAF+, E2: DTDAF+•/DTDAF2+). The introduction of a chain bridging the two thiazole rings, e.g., 2, hinders any rotation upon oxidation and, in this case, the potential difference (ΔE) is enlarged, due to increased electronic interactions in the oxidized species [4,5]. We recently showed that the introduction of a bis oxaethyl chain to bridge the two thiazole cores could modulate the redox properties of DTDAF (3 R1 = Me and R2 = COMe) as the presence of a metal cation such as Li+ accounts for the stabilization of the cation radical species by maintaining the thiazole cores in the same plane upon complexation [6]. Herein we present a novel route to DTDAF with longer oxaethyl bridging chains 4a–4c and the effect of the length of this bridging chain on the redox properties of the donor. In addition, we report also preliminary electrochemical investigations of alkali metal binding effect on their redox behaviour.

2 Results and discussion
In our precedent approach, used to prepare N,N′-bridged DTDAF 3 (Scheme 1), we started from (2,2′-(ethylenedioxy)bis(ethylamine) and then realized, at each extremity of this alkyl chain, the synthesis of bis(thiazoline-2-selones) and bisthiazolium salts, both precursors of DTDAF core [6]. Moreover, we also noticed that the coupling of these bis(thiazoline-2-selones) in the presence of P(OEt)3 afforded a DTDAF, resulting from an intermolecular coupling rather than from an intramolecular coupling as in the case of bisthiazolium salt in basic medium.
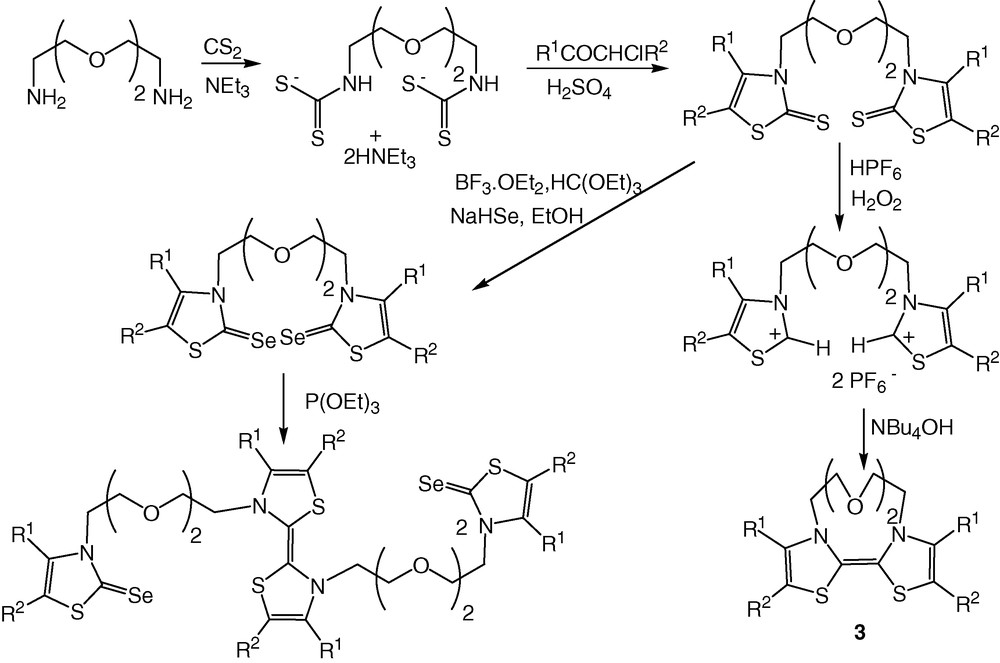
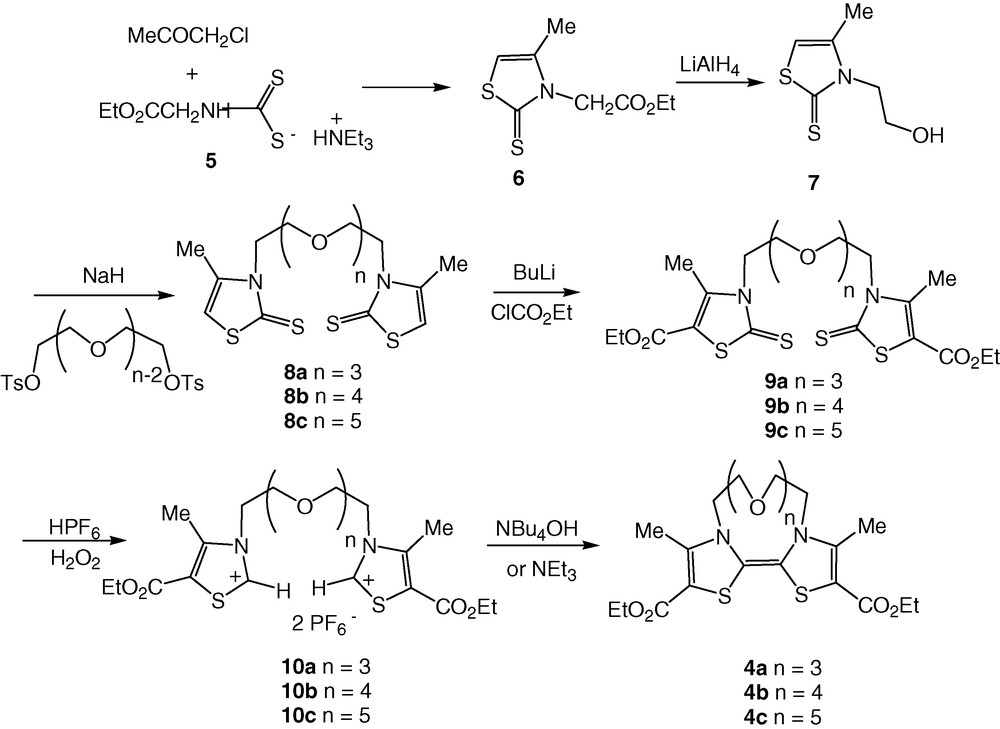
Therefore, we decided herein to investigate the formation of N,N′-bridged DTDAF 4a–4c only from the basic coupling of bisthiazolium salts and, in order to introduce the oxaethyl bridging chain of varied length, we modified our synthetic strategy. The chemical pathway we used for the synthesis of DTDAF 4a–4c is depicted in Scheme 2. The general strategy consists this time in forming the thiazole core first, substituted on the nitrogen by a hydroxyethyl group, and then linking two of them through the poly(oxaethyl) chain. Our starting material is the dithiocarbamate salt 5 obtained by simply mixing glycine ethyl ester hydrochloride with carbon disulfide and triethylamine [7]. Alkylation of this salt with chloroacetone followed by cyclisation and dehydration, in acidic medium, yields thiazoline-2-thione 6 in 73% yield. The reduction of the ester group was performed using LiAlH4 as the reducing agent and afforded thiazoline-2-thione 7 in 90% yield. In order to introduce an oxaethyl chain, we generated the alkoxide ion from 7 with sodium hydride in dry THF and added in the medium half equivalent of either the di-, tri- or tetra(ethylene glycol)di(p-tosylate). Bisthiazoline-2-thiones 8a–8c are isolated after column chromatography. As DTDAFs are highly oxygen-sensitive [8], we prepared the precursors of the donor core substituted with electron withdrawing groups such as the ethoxycarbonyl one, in order to decrease the overall donor ability of the DTDAF, to stabilize them against oxidative rearrangements and to facilitate the electrochemical study. This was realized by a sequence of lithiation/alkylation with BuLi and ethyl chloroformate. Using hexafluorophosphoric acid and hydrogen peroxide, bis(thiazoline-2-thiones) 9a–9c were quantitatively converted into the bisthiazolium salts 10a–10c. The coupling of these salts in basic medium afforded the desired crown ether annelated DTDAF 4a–4c.
In order to avoid any uncontrolled oxidation of the donor core, as DTDAFs are known to lead upon air exposure to various structures of oxygen-containing products, [8] we performed cyclic voltammetry experiments directly in the reaction mixture where the donor was formed under inert atmosphere. The oxidation peak potentials of the donors are collected in Table 1 together with the data of N,N′-ethylene bridged DTDAF 2 (R1 = Me and R2 = CO2Et) and the N,N′-bis(oxaethyl) DTDAF 3 (R1 = Me and R2 = CO2Et) for comparison [5,6]. Note that all the donors are substituted with the same exocyclic substituents. Depending on the length of the crown ether, the voltammogram exhibits either two monoelectronic processes (3, 4a) or one bielectronic process 4b–4c. If we compare the oxidation potentials of 2, 3, and 4a, which present two reversible monoelectronic waves, one can observe that the difference between the two oxidation potentials (ΔE = Epa2 – Epa1) decreases as the length of the N,N′-bridging chain increases (Fig. 1). Indeed, with an ethylene bridge the two thiazole rings are coplanar and remain coplanar upon oxidation (ΔE = 550 mV), while with a longer chain, important structural changes can occur concerted with the electron transfer and therefore, due to reduced electronic interaction, a ΔE of about 200 mV is observed. In the case of DTDAF 4b and 4c, only one reversible bielectronic oxidation wave is observed, corresponding to the oxidation of the two thiazole cores simultaneously (Fig. 1). It is worth noting that similar trends have already been reported by Thummel et al. while studying the effect of the N,N′-bridge length, from two to four carbons, on the redox behaviour of the DTDAFs [9]. Indeed, they found that ΔE decreases when the length increases until reaching only one single wave for the butyl side chain. This modification could be assigned to the fact that the bulkiest side chain would induce a distortion of the donor core in the neutral state and the donor would be less conjugated, modifying the energetic stabilities of cation radical species. Therefore, upon oxidation, the dicationic state is reached easily.
Oxidation peak potentials of the donors formed in the medium after the chemical coupling with aNBu4OH bNEt3, E in V vs SCE, Pt working electrode with 0.1-M n-Bu4NPF6 in CH3CN, scanning rate 0.1 V s–1, ΔEp = Epa2 – Epa1
| Epa 1 | Epa 2 | ΔEp (mV) | Epa 1 | Epa 2 | ΔEp (mV) | ||
| 2 5 | –0.27 | 0.28 | 550 | ||||
| 3 a,6 | –0.21 | 0.02 | 230 | 3–Li+ | –0.205 | 0.05 | 255 |
| 4a a | –0.13 | 0.08 | 210 | ||||
| 4b a,b | –0.15 | — | 4b–Li+ | –0.13 | |||
| 4c b | –0.13 | — | 4c–Na+ | –0.065 | |||
| 4c–K+ | –0.09 |

Cyclic voltammograms for DTDAFs 2 (dotted line) 3 (dashed line) and 4b (solid line), Pt working electrode with 0.1 M Bu4NPF6 in CH3CN.
As observed with the crown-ether-annelated tetrathiafulvalenes (TTF), the cation binding effect can be detected by cyclic voltammetry via the shift of the redox processes due to electrostatic interaction and proximity of the crown-ether-bound cation with the donor core [10]. Therefore, we studied the redox properties of these DTDAFs in the presence of alkali metal ions (Li+, Na+, and K+). First we analysed 3(R1 = Me and R2 = CO2Et), and we observed the same trends as previously observed on DTDAF 3 bearing an acetyl substituent (R2 = COMe) instead of an ethoxycarbonyl [6]. Indeed, the largest positive shift of the oxidation potential was obtained in the presence of one equivalent of Li+, with a more pronounced shift on the second oxidation process than on the first one (+5 mV on Epa1 and +30 mV on Epa2) and no shift has been observed on the reduction waves. This indicates that the ethoxycarbonyl substituents do not modify the overall effect of the side-chain complexation. This effect on both oxidation processes is due to electrostatic repulsion with the bound metal ion, indicating that the cation was bound to the side chain during the two oxidation processes and then expelled out of the cavity, as no shift was detected on the reduction waves.
Then we analysed the redox behaviour of 4a–c in the presence of alkali metal ions (Li+, Na+, and K+). The most significant results are collected in Table 1. Only modifications on the cyclic voltammograms are observed for the DTDAFs 4b in the presence of Li+ and 4c in the presence of Na+ and K+. In all these cases, a positive shift of the oxidation potentials was observed and the maximum potential shift was reached after the addition of 1 equiv of cation, while for the reduction processes no modification was observed. The fact that only the oxidation wave leading to the dication is shifted tends to indicate that the metal ion is complexed with the crown-annelated DTDAFs 4b and 4c, but as the reduction waves are not affected, the metal ion was expelled out of the cavity upon oxidation to the dication. The largest positive shift for 4c was reached with the sodium ion (65 mV), and 40 mV for the potassium ion, probably due to the better correlation of the geometry of the cavity and the metal ion. The fact that the presence of the alkali cations modifies only the oxidation potentials suggests that complexation occurs, but does not modify the conformation of the neutral crown-annelated DTDAF; it could be for example a planarisation of the donor core, which would induce a splitting of the bielectronic wave into two monoelectronic waves.
3 Conclusion
A series of crown-ether annelated DTDAF has been synthesized with a poly(oxaethyl) bridging chain of various lengths. Analysis of the redox behaviour by cyclic voltammetry shows that molecules with the longest poly(oxaethyl) chain (n = 4,5) exhibit one reversible bielectronic transfer, while molecules with the shorter one (n = 2,3) exhibit two reversible monoelectronic transfers. Preliminary complexation experiments show that the largest positive shift upon complexation (65 mV) is obtained with n = 5 and the sodium cation. Further work is underway to study other cations than alkali metal ions and their effect on the redox processes of these crown ether annelated DTDAFs.
4 Experimental
4.1 General remarks
THF was distilled from sodium-benzophenone prior to use. Chromatography experiments were performed using silica gel Merck 60 (70–260 mesh). 1H NMR spectra were recorded at 300 M Hz and 13C NMR spectra at 75 M Hz with tetramethylsilane as internal reference. Mass spectra were carried out at the ‘Centre de mesures physiques de l’Ouest’, Rennes, France. Melting points were measured using a Kofler hot-stage apparatus and are uncorrected. Elemental analysis results were performed at the ‘Laboratoire central de microanalyse du CNRS’, Lyons, France.
4.2 Synthesis of dithiocarbamate salt (5)
Glycine ethyl ester hydrochloride (4.17 g, 30 mmol) was suspended in triethylamine (50 ml) at 0 °C and 50 ml of carbon disulfide were added. The mixture was allowed to reach room temperature and was stirred for 24 h. After addition of 200 ml of ether, a white precipitate was formed, filtered off and washed with dry ether. Carbamate salt 5 was used without further purification (quantitative yield, very hygroscopic), m.p. 71 °C, 1HNMR (CDCl3): δ 1.28 (t, 3H, JCH–CH = 7.1 Hz, CH2CH3), 1.41 (t, 9H, JCH–CH = 7.3 Hz, N(CH2CH3)3), 3.20 (q, 6H, JCH–CH = 7.3 Hz, N(CH2CH3)3), 3.51 (s, 1H, NH), 4.21 (q, 2H, JCH–CH = 7.1 Hz, CH2CH3), 4.41(d, 2H, JNH–CH = 4.8 Hz, CH2NH).
4.3 Ethyl (4-methyl-2-thioxo-1,3-thiazol-3(2H)-yl)acetate (6)
Dithiocarbamate salt 5 (starting from 30 mmol of GlyOEt·HCl) was suspended in CH3CN (100 ml) and 2-chloroaceton (2.4 ml, 30 mmol) was added dropwise. The reaction mixture was stirred at room temperature for 24 h. The reaction mixture was concentrated under reduced pressure and 1 ml of sulphuric acid (98%) was added dropwise. After 15 min of stirring, 100 ml of water was added. A white precipitate was formed and filtered off. The filtrate was extracted with CH2Cl2 (3 × 60 ml). The organic layers were combined, washed with water (3 × 100 ml) and dried over Na2SO4. The solvent was evaporated, solid residue remained, which was combined with the crystalline product and recrystallized from a mixture of ethanol and water (yield, 73%), m.p. 94 °C, 1HNMR (CDCl3): δ 1.33 (t, 3H, JCH–CH = 7.1 Hz, CH2CH3), 2.26 (d, 4J = 1.17 Hz, 3H, CH3), 4.29 (q, 2H, JCH–CH = 7.1 Hz, CH2CH3), 5.02 (s, 2H, CH2), 6.32 (d, 1H, 4J = 1.17 Hz, CH). 13C NMR (CDCl3): δ 14.11, 15.31, 47.65, 62.11, 105.88, 139.43, 166.49, 189.34. Anal. calcd for C8H11NS2O2: C, 44.22; H, 5.10; N, 6.45; S, 29.51. Found: C, 44.20; H, 5.01; N, 6.49; S, 29.51.
4.4 3-(2-hydroxyethyl)-4-methyl-1,3-thiazol-2(3H)-thione (7)
LiAlH4 (1.15 g, 30 mmol) was suspended in 25 ml of dry THF. The suspension was degassed and cooled to –20 °C. Ethyl (2-thioxo-1,3-thiazol-3-yl)acetate 6 (4.3 g, 20 mmol) was dissolved in 50 ml of dry THF and added dropwise to the suspension of LiAlH4. The reaction mixture was stirred for additional 30 min at –20 °C. The excess of LiAlH4 was hydrolysed by addition of a mixture of THF (40 ml) and water (40 ml). After acidification of the medium, with 4 N HCl, the solution was extracted with ethyl acetate (3 × 50 ml). The combined organic layers were washed with water (100 ml) and brine (100 ml), dried over Na2SO4, solvent was evaporated under vacuo to give a colourless oily residue, which was purified by distillation and crystallized upon cooling (yield, 89%; white powder), m.p.: 68 °C, 1H NMR (DMSO-d6): δ 2.34 (d, 3H, 4J = 1.1 Hz, CH3), 3.72 (q, 2H, J = 5.4 Hz, CH2), 4.19 (t, 2H, JCH–CH = 5.6 Hz, CH2), 5.04 (t, 1H, J = 5.2 Hz, OH), 6.73 (d, 1H, 4J = 1.1 Hz, H). 13C NMR (DMSO-d6): δ 15.56, 49.62, 57.70, 106.30, 141.98, 186.96. Anal. calcd for C6H9NS2O: C, 41.12; H, 5.18; N, 7.99; S, 36.58. Found: C, 40.86; H, 5.03; N, 7.94; S, 36.92.
4.5 General procedure for the synthesis of 8a–c
To a solution of 7 (2.63 g, 15 mmol) in dry THF was added 1.5 equiv of sodium hydride (0.54 g, 22.5 mmol) under inert atmosphere. After stirring for 1 h at room temperature, a solution of di, tri or tetra(ethylene glycol)di-p-tosylate (7.5 mmol) in dry THF was added to the medium. The reaction mixture was heated under reflux for 48 h and then water was added. Extraction with dichloromethane, drying over Na2SO4 and evaporation of the solvent lead to an oil. Chromatography on silica gel eluting with dichloromethane: ether (50:50) gave bisthiazoline thiones 8 as pale yellow oils.
4.5.1 3,3′-(oxobis(ethane-2,1-diyloxyethane-2,1-diyl))bis(4-methyl-1,3-thiazole-2(3H)-thione) 8a
Yield: 52%; 1H NMR (CDCl3): δ 2.34 (d, 6H, 4J = 0.8 Hz, CH3), 3.53 (s, 8H, OCH2CH2O), 3.86 (t, 4H, OCH2CH2N, 3J = 5.1 Hz), 4.36 (t, 4H, OCH2CH2N, 3J = 5.1 Hz), 6.24 (d, 4J = 0.8 Hz, 2H, =CH); 13C NMR (CDCl3): δ 16.15, 47.87, 67.98, 70.76, 71.00, 106.17, 141.67, 188.19; anal. calcd for C16H24N2O3S4: C, 45.69; H, 5.75; N, 6.66; S, 30.49. Found C, 45.68; H, 5.77; N, 6.67; S, 30.62.
4.5.2 3,3′-(3,6,9,12-tetraoxatetradecane-1,14-diyl))bis(4-methyl-1,3-thiazole-2(3H)-thione) 8b
Yield: 72%; 1H NMR (CDCl3): δ 2.28 (d, 6H, 4J = 1.1 Hz, CH3), 3.48 (s, 12H, OCH2CH2O), 3.77 (t, 4H, OCH2CH2N, 3J = 5.1 Hz), 4.28 (t, 4H, OCH2CH2N, 3J = 5.1 Hz), 6.21 (d, 2H, 4J = 1.1 Hz, =CH); 13C NMR (CDCl3): δ 16.11, 47.85, 67.95, 70.81, 70.93, 71.28, 106.88, 141.9 188.11.
4.5.3 3,3′-(3,6,9,12,15-pentaoxaheptadecane-1,17-diyl)bis(4-methyl-1,3-thiazole-2(3H)-thione) 8c
Yield: 60%; 1H NMR (CDCl3): δ 2.35 (d, 6H, 4J = 1.14 Hz, CH3), 3.57 (s, 8H, OCH2CH2O), 3.60 (s, 8H, OCH2CH2O), 3.85 (t, 4H, OCH2CH2N, 3J = 5.1 Hz), 4.36 (t, 4H, OCH2CH2N, 3J = 5.1 Hz), 6.21 (d, 2H, 4J = 1.14 Hz, =CH); 13C NMR (CDCl3): δ 15.47, 47.22, 67.34, 70.12, 70.20, 70.23, 70.30, 105.39, 140.98, 187.58. Anal. calcd for C20H32N2O5S4 : C, 47.22; H, 6.34; N, 5.51. Found C, 46.80; H, 6.34; N, 5.45.
4.6 General procedure for the synthesis of 9a–c
To a solution of the N,N′-bridged bis (thiazoline-2-thione) 8 (2 mmol) in dry THF (30 ml) was added n-BuLi (3.2 ml, 5 mmol, from a 1.6-M solution in hexane) at –80 °C under inert atmosphere. After stirring for 1.5 h, ethyl chloroformate (0.38 ml, 4 mmol) was added and the solution was slowly allowed to reach room temperature and stirred for an additional 3 h. Solvent was evaporated and CH2Cl2 (50 ml) was added to the resulting oil. The organic phase was washed several times with water (3 × 50 ml), dried over Na2SO4, and the solvent was evaporated. Column chromatography of the residue on silica gel column with CH2Cl2–Et2O 80:20 as eluent afforded bisthiazoline thiones 9 as colourless oils.
4.6.1 Diethyl 3,3′-(oxybis(ethane-2,1-diyloxyethane-2,1-diyl))bis(4-methyl-2-thioxo-2,3-dihydro-1,3-thiazole-5-carboxylate 9a
Yield: 37%; 1H NMR (CDCl3) δ 1.34 (t, 6H, CH3), 2.74 (s, 6H, CH3), 3.53 (s, 8H, OCH2CH2O), 3.85 (t, 4H, OCH2CH2N, 3J = 5.1 Hz), 4.30 (q, 4H, CH2), 4.42 (t, 4H, OCH2CH.2N, 3J = 5.1 Hz); 13C NMR (CDCl3) δ 14.66, 14.75, 48.10, 61.90, 67.95, 70.84, 71.00, 106.07, 150.07, 160.48, 188.99; anal. calcd for C22H32N2O7S4 : C, 46.79 ; H, 5.71 ; N, 4.96. Found C, 46.33; H, 5.91; N, 4.57.
4.6.2 Diethyl 3,3′-(3,6,9,12-tetraoxatetradecane-1,14-diyl))bis(4-methyl-2-thioxo-2,3-dihydro-1,3-thiazole-5-carboxylate 9b
Yield 31%; 1H NMR (CDCl3) δ 1.37 (t, 6H, CH3), 2.77 (s, 6H, CH3), 3.59 (s, 12H, OCH2CH2O), 3.89 (t, 4H, OCH2CH2N, 3J = 5.1 Hz), 4.32 (q, 4H, CH2), 4.45 (t, 4H, OCH2CH2N, 3J = 5.1 Hz); 13C NMR (CDCl3) δ 14.61, 14.70, 48.09, 61.80, 67.92, 70.78, 70.88, 70.96, 111.69, 150.07, 160.38, 188.88; Anal. calcd for C24H36N2O8S4 : C, 47.35; H, 5.96 ; N, 4.60; S, 21.07. Found C, 47.22; H, 6.06; N, 4.44; S, 21.26.
4.6.3 Diethyl 3,3′-(3,6,9,12,15-pentaoxaheptadecane-1,17-diyl)bis(4-methyl-2-thioxo-2,3-dihydro-1,3-thiazole-5-carboxylate 9c
Yield: 15%; 1H NMR (CDCl3) δ 1.26 (t, 6H, CH3), 2.66 (s, 6H, CH3), 3.49 (s, 8H, OCH2CH2O), 3.52 (s, 8H, OCH2CH2O), 3.78(t, 4H, OCH2CH2N, 3J = 5.1 Hz), 4.22 (q, 4H, CH2), 4.34 (t, 4H, OCH2CH2N, 3J = 5.1 Hz); 13C NMR (CDCl3) δ 13.23, 13.33, 46.74, 60.46, 66.61, 69.46, 69.57, 69.65, 110.46, 148.45, 159.12, 187.72; Anal. calcd for C26H40N2O9S4 : C, 47.83 ; H, 6.18; N, 4.29. Found C, 47.87; H, 6.33; N, 3.88.
4.7 General procedure for the synthesis of 10a–c
To a suspension of bis thiazoline thione 9 (1 mmol) in 10 ml of acetone at 0 °C was added, under stirring, HPF6 (2 mmol, 60% solution in water) and H2O2 (16 mmol, 35% solution in water). The reaction mixture was warmed slowly at 50 °C until the formation of a homogeneous solution. After addition of water and extraction with CH2Cl2 (2 × 40 ml), the combined organic phases were washed with water and concentrated in vacuo to afford 10a–c as colourless oils, which were used in the next step without further purification.
4.7.1 3,3′-[oxybis(ethane-2,1-diyloxyethane-2,1-diyl)]bis[5-(ethoxycarbonyl)-4-methyl-2,3-dihydro-1,3-thiazol-2-ylium]dihexafluorophosphate 10a
Yield: 90%; 1H NMR ((CD3)2CO) δ 1.41 (t, 6H, CH3), 2.99 (s, 6H, CH3), 3.59 (m, 4H, OCH2CH2O), 3.68 (m, 4H, OCH2CH2O), 4.07 (t, 4H, OCH2CH2N, 3J = 4.7 Hz), 4.49 (q, 4H, OCH2CH3), 4.88 (t, 4H, NCH2CH2O, 3J = 4.7 Hz), 10.19(s, 2H, CH); 13C NMR ((CD3)2CO) δ 12.35, 13.42, 53.47, 63.15, 67.54, 69.93, 70.24, 126.62, 152.45, 158.85, 160.99.
4.7.2 3,3′-(3,6,9,12-tetraoxatetradecane-1,14-diyl)bis[5-(ethoxycarbonyl)-4-methyl-2,3-dihydro-1,3-thiazol-2-ylium] dihexafluorophosphate 10b
Yield 73%; 1H NMR ((CD3)2CO) δ 1.41 (t, 6H, CH3), 2.99 (s, 6H, CH3), 3.61 (s, 4H, OCH2CH2O), 3.62 (m, 4H, OCH2CH2O), 3.70 (m, 4H, OCH2CH2O), 4.07 (t, 4H, OCH2CH2N, 3J = 4.7 Hz), 4.49 (q, 4H, CH2), 4.88 (t, 4H, OCH2CH2N, 3J = 4.7 Hz), 10.24 (s,2H, CH); 13C NMR ((CD3)2CO) δ 12.34, 13.42, 53.40, 63.14, 67.38, 69.87, 70.09, 70.21, 126.61, 152.38, 158.87, 161.23. HRMS calcd for (C24H38N2O8S2)2+ m/z: 273.1034. Found 273.1037.
4.7.3 3,3′-(3,6,9,12,15-pentaoxaheptadecane-1,17-diyl)bis[5-(ethoxycarbonyl)-4-methyl-2,3-dihydro-1,3-thiazol-2-ylium] dihexafluorophosphate 10c
Yield: 70%; 1H NMR ((CD3)2CO) δ 1.41 (t, 6H, CH3), 2.99 (s, 6H, CH3), 3.63 (m, 16H, OCH2CH2O), 4.05 (t, 4H, OCH2CH2N, 3J = 4.7 Hz), 4.47 (q, 4H, CH2), 4.87 (t, 4H, OCH2CH2N, 3J = 4.7 Hz), 10.27 (s,2H, CH); 13C NMR ((CD3)2CO) δ 14.15, 15.23, 55.21, 64.91, 68.18, 71.68, 71.79, 71.86, 71.93, 129.34, 154.14, 159.93, 163.25.
4.8 General procedure for the preparation of DTDAF 4 and in situ electrochemical investigations
To a 0.1-M solution of tetrabutylammonium hexafluorophosphate in 20 ml of CH3CN, bisthiazolium salt 10 (0.05 mmol) was added. The reaction mixture was degassed and NBu4OH (0.1 mmol, 40% solution in water) or NEt3 (0.1 mmol) was added under nitrogen. The solution turned immediately orange-red indicating the formation of the donor core in the medium. The CV of the solution was then recorded.