

1 Introduction
The present paper concerns the ability of Cu+ ions in zeolite to activate some organic molecules: alkenes, alkynes, aromatic hydrocarbons and ketones. Since the early reports by Iwamoto et al. [1–4] on the activity of CuZSM-5 in the decomposition of nitrogen oxides, copper containing zeolites attracted a great deal of attention. DFT calculations showed [5–8] that the high activity of Cu+ in CuZSM-5 could be related to the high energy of the HOMO orbital of Cu+ in MFI and to the π-back donation of electrons from the d orbitals of Cu to π* antibonding orbitals of NO, resulting in a distinct weakening and dissociation of the N=O bond. The role of zeolite was the partial neutralization of the electrical charge of Cu+: the charge decreased from +1 to +0.3 when Cu+ was placed in a cluster simulating a fragment of the MFI structure, and therefore, in an increase of the HOMO energy. According to Goursot et al. [9], zeolitic framework acts as a reservoir of electrons—the negative charge transferred to NO comes mostly from the framework with only a small variation in the charge on the Cu+ ion.
Apart from of the activity in the ‘denox’ reaction, Cu-containing zeolites were also found to be active catalysts in some reactions of organic molecules [10–23]. It was recently reported that CuMCM-41 is active in n-butenes isomerization to isobutene, known to be an acid catalyzed reaction. In butenes isomerization CuMCM-41 was found to be more active than HMCM-41, despite its lower Brønsted acidity, indicating the important role of Cu+ ions as active sites in this reaction. The activity of Cu-containing faujasites in Diels–Alder cyclodimerization of 1–3 butadiene to vinylcyclohexene was reported by Piffer et al. [23] Cu–zeolites are also active in some reactions of alkene oxidation [13–16,18–22].
Since most of the aforementioned reactions concern alkenes, we undertook IR studies of the activation of the alkene double bond by Cu+ and of the triple C≡C bond in acetylene. We studied also the activation of C–C bond in benzene and of C=O bond in acetone. It was expected that Cu+, which activated double bond in NO by π-back donation, might also activate multiple bond in other molecules following the similar mechanism.
We performed the IR studies and quantum chemical DFT calculations concerning the adsorption of: but-1-ene, cis-but-2-ene, trans-but-2-ene, propene, ethene, acetylene, benzene, and acetone on Cu+ sites in zeolites CuX, CuY, and CuZSM-5 zeolites. The adsorption was done at room temperature in most IR studies. As CuZSM-5 besides Cu+ sites contains strongly acidic protonic sites, which may catalyze but-1-ene isomerization and oligomerization of all the alkenes, they were adsorbed at low temperature (170 K) to slow down these undesirable reactions.
2 Experimental
Zeolites CuX, CuY and CuZSM-5 zeolites were prepared from zeolite NaX (Si/Al = 1.3) obtained from the Department of Chemical Technology, Jagiellonian University. Zeolite NaY and NaZSM-5 (Si/Al = 2.55 and 35, respectively) from the Institute of Industrial Chemistry (Warsaw) were treated with Cu(CH3COO)2 solution at 350 K. Following ion exchange the samples were washed with distilled water and dried in air at 370 K. The exchange degrees (Cu/Al) were 0.43, 0.31 and 0.45 for CuX, CuY, and CuZSM-5, respectively. We also used silicalite synthesized at Department of Chemical Technology, Jagiellonian University. In some experiments, HY and HZSM-5 were used. They were obtained by the ionic exchange from parent Na-forms with NH4Cl, what was followed by the activation in situ in IR cell at 730 K (NaHY) and 870 K (HZSM-5). The exchange degrees (H/Al) were 77% and 95% as received.
But-1-ene, cis-but-2-ene, trans-but-2-ene, and propene (Fluka – 99.9%), ethene (UCAR – 99.5%), acetylene (Messer – 99.6%), benzene (Aldrich – analytical grade) and acetone (Aldrich – analytical grade) were used in IR experiments. As acetylene stored in a cylinder contains some amount of acetone, it was purified by freeze-thaw technique, until disappearance of the band of carbonyl group (ca. 1720 cm–1).
For IR studies the Cu–zeolites were pressed into thin wafers and activated in situ in IR cell at 730 K at vacuum for 1 h. IR spectra were recorded by BRUKER 48 PC spectrometer equipped with an MCT detector. Spectral resolution was 2 cm–1.
2.1 Quantum chemical DFT calculations
DFT calculations were carried out for clusters models by Dmol Software of MSI© (Ref. [24]). Dmol code is the implementation of numerical scheme for solving Kohn–Sham equations. We have chosen standard calculation parameters, e.g. local VWN exchange-correlation potential and numerical DNP basis set. Inner core orbitals were frozen during calculations. This choice was promoted by the compromise between computational efficiency and expected accuracy. The properties to be discussed here are geometrical parameters, charge distribution, Mayer bond orders.
Preliminary model of a copper site was cut off the MFI structure taken from MSI databases included in the software. It comprised 7T atoms arranged into two fused 5T rings forming a 6T ring, which simulated framework environment for copper cations. The Cu+ cation was than placed in the center of 6T ring and its position was optimized with constrained coordinates of protons terminating the cluster.
The interacting system was constructed from a cluster and the appropriate molecule: but-1-ene, cis-but-2-ene, trans-but-2-ene, propene ethene, acetylene, benzene and acetone. The composed system was then optimized and properties in question were calculated from population and vibrational analyses.
3 Results and discussion
3.1 The electron acceptor properties of molecules and their effect on bond activation
The observed by us [5–8] change of charge on copper after placing the cation in a zeolite framework suggests that the embedding may strongly influence its π-donor properties. It shows also the function of a zeolite as an electron reservoir, enhancing the ability of Cu+ to π-backdonation.
However, the phenomenon of flowing electrons from a site to an interacting molecule may be also moderated by electron affinity of the molecule. The electron affinity reveals ability of a molecule to be an acceptor of π-back donation. Thus we have calculated by DFT method (as described above) the values of electron affinities for a series of alkenes: ethane, propene, but-1-ene, cis-but-2-ene, trans-but-2-ene, acetylene, benzene and acetone, as well as for CO and N2, which were used in our previous study [8] as probe molecules. The electron affinities were the difference between total energies of anions and neutral molecules.
The obtained values are shown in Table 1. One can expect that the smaller energy loss (higher effective electron affinity) the more electrons will be gained by the molecules and the larger will be the activation. Calculated energy loss for alkenes, acetylene, and benzene is ca. 1.4 eV, what is more than for acetone and NO but less than for CO and N2. Therefore alkenes, acetylene, and benzene will be relatively good acceptors of electrons (better than CO and N2, but still not as good as NO). This suggests that they could be activated by π-back donation while interacting with Cu+ site in zeolites, which is well-known from a great ability to π-back donation. Acetone is better electron acceptor (electron affinity = 0.8 eV) than mentioned above hydrocarbons.
The values of electron affinities (EA) of molecules studied (kJ mol–1)
| Molecule | EA |
| NO | 0.5 |
| CO | 1.9 |
| N2 | 2.1 |
| Ethene | 1.7 |
| Propene | 1.5 |
| But-1-ene | 1.4 |
| Cis-but-2-ene | 1.7 |
| Trans-but-2-ene | 1.5 |
| Acetylene | 1.5 |
| Benzene | 1.3 |
| Acetone | 0.8 |
The extent of the activation of the appropriate bond in a molecule after accepting the electron due to π-backdonation depends obviously on the character of the accepting molecular orbital. While in alkenes or acetylene this orbital is strongly antibonding, in benzene molecule is only weakly antibonding or even non-bonding. Thus the activation of C=C bond in alkenes or C≡C bond in acetylene should be much more pronounced than the activation of benzene. In benzene only very weak activation of C–C bond is expected.
3.2 IR studies of alkenes sorption
3.2.1 The interaction of double bond with Cu+ ions in zeolites
According to the results described in details in our previous work [25,26], the interaction of alkenes with Cu+ in zeolites resulted in a distinct weakening of C=C bond, and is some perturbation of C-H bonds in =CH2 and =CH groups near to the double bond.
The weakening of C=C double bond is well seen in Figs. 1 and 2 in which the spectra of but-1-ene, cis-but-2-ene, trans-but-2-ene, propene and ethene adsorbed in CuX zeolite are shown. In the case of ethene, the red shift of C=C band (ΔνC=C) was 78 cm–1 whereas for other alkenes ΔνC=C was 105–115 cm–1. In the case of ethene, C=C IR band which was IR inactive in free molecule became active, when ethene interacted with Cu+ (Fig. 2). This result indicated that ethene molecule lost its symmetry when interacting with Cu+. Such a loss of symmetry was, however, not observed in the case of trans-but-2-ene – no C=C band was seen when trans-but-2-ene was sorbed on CuX (Fig. 1).
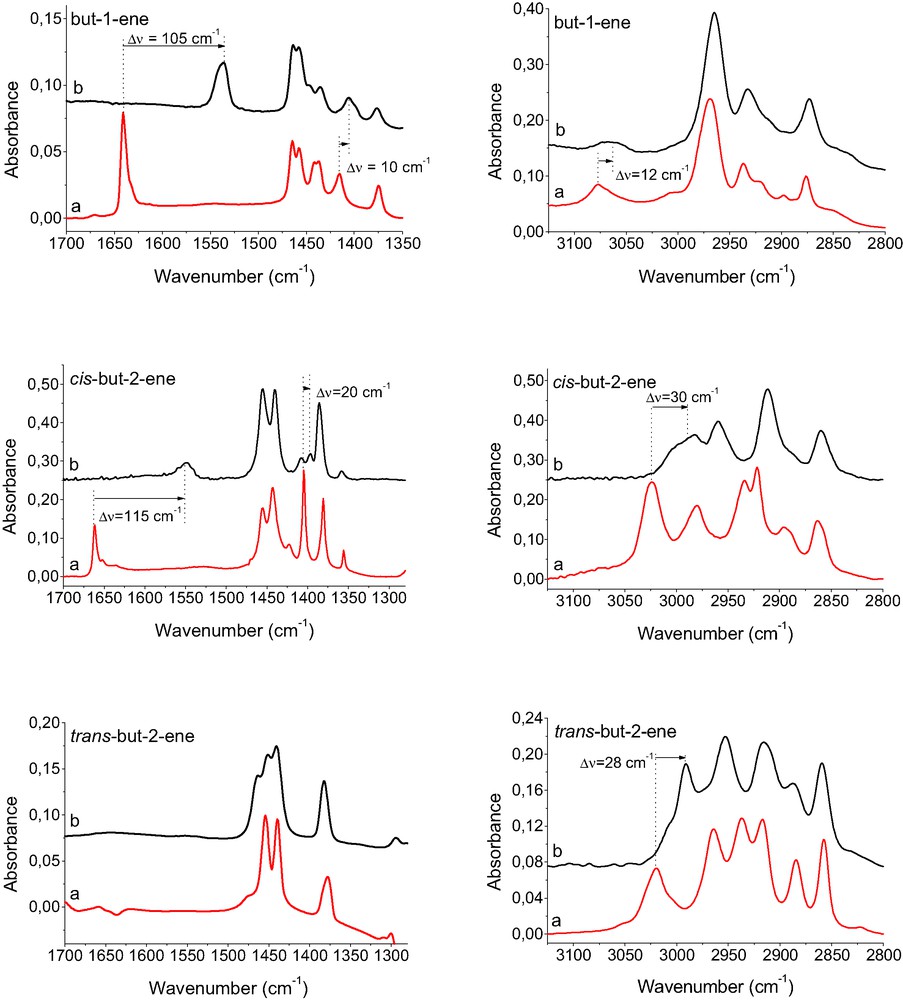
IR spectra of but-1-ene, cis-but-2-ene and trans-but-2-ene physisorbed in silicalite (a) and sorbed in CuX (b).
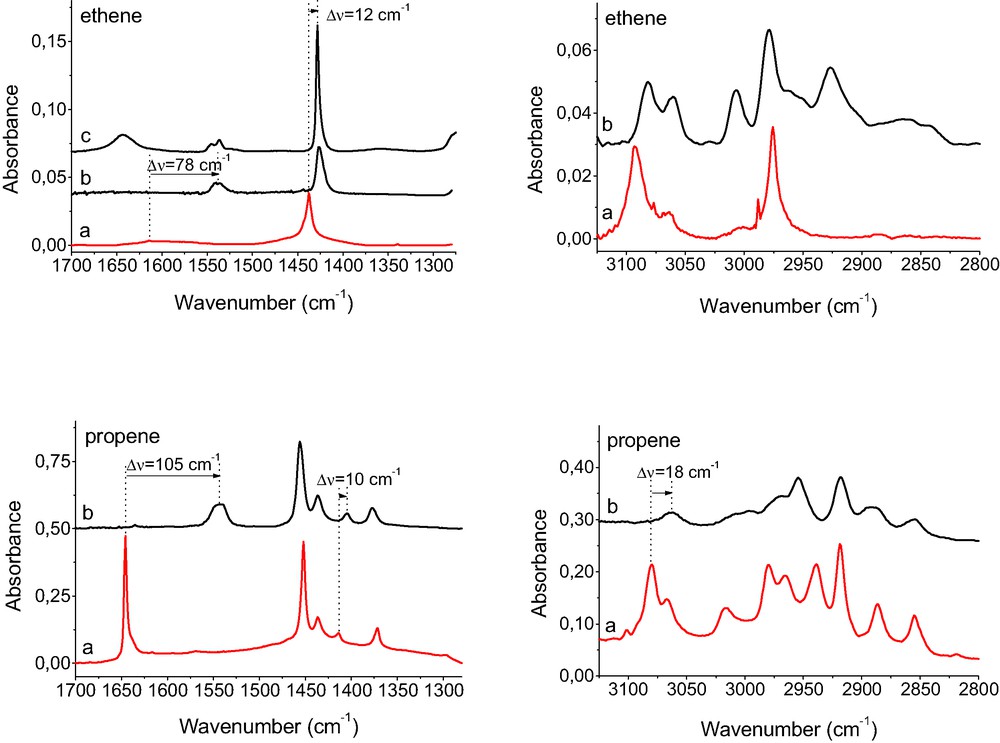
IR spectra of propene and ethene physisorbed in silicalite (a) as well as sorbed in CuX (b) and in CuY (c).
It is very possible, that this weakening of C=C bond was a result of π back donation of d electrons of Cu+ ions to π* antibonding orbitals of alkenes. As mentioned in the Introduction, quantum chemical DFT calculations show that the location of Cu+ in cationic positions of ZSM-5 results in a partial neutralization of its electrical charge (a decrease from +1 to +0.3) and in a distinct increase of HOMO energy (from –14.115 eV for free Cu+ to –5.170 or –5.317 eV), depending on cation location, for Cu+ in the clusters denoting an increase of electrodonor properties of Cu+ [7,8].
The analysis of the spectra shown in Figs. 1 and 2 showed that not only the C=C bond was affected by the interaction with Cu+ but also the =CH2 and =C–H bonds were perturbed. The IR bands of =CH2 and =C–H stretching (3000–3100 cm–1) were red shifted by 12 cm–1 (but-1-ene) to 30 cm–1 (cis-but-2-ene). The bands of =CH2 scissoring (1400–1450 cm–1) were red shifted by 10–12 cm–1 (for but-1-ene, propene, and ethene) and the band of =C–H bending (1400 cm–1) in cis-but-2-ene was shifted by 20 cm–1. The vibrations of CH3 and CH2 groups, more distant from the double bond, were less perturbed. Similar results: distinct weakening of C=C bonds and some perturbation of =CH2 and =C–H bonds were also observed when alkenes were sorbed in CuY and CuZSM-5 (spectra not shown). In the case of ethene sorbed in CuY (Fig. 2), the band of C=C vibration is split into two maxima at: 1535 and 1546 cm–1 (Δν = 81 and 70 cm–1). Similar results were obtained by Hűbner and Roduner [27], who reported distinct red shift and splitting of both C=C stretching and =CH2 scissoring bands in ethene interacting with Cu+ in CuY. The values of ΔνC=C = 78 and 88 cm–1, reported by Hűbner and Roduner [27] are very close to those obtained in our study (70 and 81 cm–1). The problem of heterogeneity of Cu+ sites will be discussed in further chapter.
3.2.2 Various loading of alkenes
The results of experiments in which various amount of alkenes were sorbed in Cu–zeolites [25,26] evidenced that all the alkenes studied, reacted with Cu+ cations in the first order, and upon the saturation of all Cu+ they interacted with OH groups, forming the hydrogen bonding. This is shown in Fig. 3A in which the spectra recorded at increasing loading of but-1-ene in CuX zeolite are given. At relatively low loading the band of 1540 cm–1 of but-1-ene interacting with Cu+ was present, whereas at higher loading a band of hydrogen bonded but-1-ene (1630 cm–1) appeared. At high loading another band at 1570 cm–1 appeared. Similar results: appearance of 1570 cm–1 band at high alkenes loading was also observed [25] when other alkenes: cis-but-2-ene or propene were sorbed in CuX, CuY, and CuZSM-5. In most cases the 1570 cm–1 band was growing, at high alkenes loading, at the expense of 1540 cm–1 band. An opposite effect: decrease of 1570 cm–1 band accompanied by an increase of 1540 cm–1 one was observed upon the desorption of alkenes from Cu–zeolites.
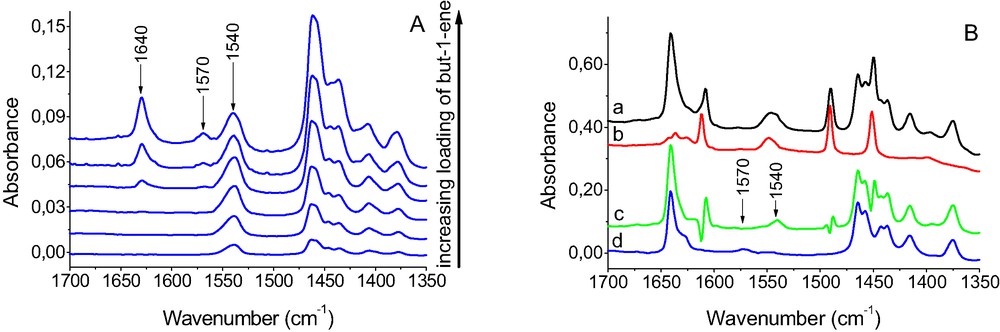
A—IR spectra of recorded upon the sorption of increasing amount of but-1-ene in CuX. B—the spectra of but-1-ene sorbed at 170 K in CuZSM-5 with preadsorbed pyridine. a—spectrum of zeolite with sorbed pyridine (sorption of pyridine at 450 K followed. by desorption at 590 K). b—bu-1-ene sorbed in zeolite with pyridine. c—difference spectrum (c = b – a). d—but-1-ene sorbed in zeolite without pyridine.
All these results suggest that the band at 1570 cm–1 may appear when two alkene molecules are bonded to one Cu+ ion. The ability of Cu+ ions to bond two NO molecules or two to three CO molecules has been reported [28–33]. The latter suggests that those sites may also bond two alkene molecules.
In CuZSM-5 zeolite, in which but-1-ene was sorbed at low temperature (170 K), 1540 and 1470 cm–1 bands appeared together [25] even at relatively low loading. It may be explained by the condensation of but-1-ene at the entrance to pores and in a high concentration of but-1-ene, which favored the bonding of two molecules by one Cu+.
The experiments in which but-1-ene was adsorbed at 170 K in CuZSM-5 with preadsorbed pyridine brought another evidence that the presence of the 1570 cm–1 band corresponded to the situation of two alkene molecules bonded to one Cu+ ion. When the excess of but-1-ene was adsorbed at 170 K on CuZSM-5, in which all Cu+ ions bonded one pyridine molecule (Fig. 3B), the only C=C band of adsorbed butene was that at 1540 cm–1 (Fig. 3B, spectrum c). The band 1570 cm–1 was absent. On the other hand 1570 cm–1 band was present when but-1-ene was sorbed at the same conditions in CuZSM-5 without pyridine (Fig. 3B, spectrum d). It seems that Cu+ ion which bonded pyridine molecule were able to bond only one alkene molecule, whereas without pyridine only some Cu+ formed 1:1 complexes with alkenes, while most of them bonded two alkene molecules.
It is not excluded that the higher C=C stretching frequency of 1570 cm–1 band (lower Δν) may be due to the fact that the charge transferred by Cu+ to alkenes is shared between two molecules.
3.2.3 Isomerization of but-1-ene bonded to Cu+ ions
Alkene molecules when sorbed in acidic zeolites (or adsorbed on other acidic catalysts) are prone to accept protons forming carbenium ions. In the case of zeolites such carbenium ions are localized inside channels and cavities, and their positive charge is stabilized by the negative charge of zeolite framework. The carbenium ions undergo transformation to more stable forms (secondary or tertiary carbenium ions) and finally alkene molecules isomerize.
It was interesting to know if the bonding of alkene molecules to Cu+ ions in zeolites makes them more or less prone to accept protons and isomerize. Brønsted acid sites, i.e. Si–OH–Al groups, which are always present in Cu–zeolites, are potential donors of protons. As the π back donation, i.e. donation of d-electrons from Cu+ to π* antibonding oritals of alkenes always takes place, a negative charge on alkene molecule appears (vide infra – Table 4). It was therefore expected that such negatively charged molecule would be more prone to accept protons than free or physisorbed molecule.
Adsorption energy Eads (kJ mol–1), elongation of the bond R (Å), decrease of Mayer bond order (b.o.), calculated and observed red shift ν (cm–1) in alkenes and acetylene interacting with Cu+ sites
| Ethene | Propene | But-1-ene | Cis-but-2-ene | Trans-but-2-ene | Acetylene | |
| Eads | 202 | 200 | 181 | 200 | 184 | 223 |
| R | +0.050 | +0.047 | +0.046 | +0.050 | +0.043 | +0.040 |
| b.o. | –0.64 | –0.60 | –0.54 | –0.51 | –0.58 | –0.76 |
| νcalc | –133 | –133 | –141 | –124 | –112 | –178 |
| νexp | –78 | –104 | –105 | –113 | –a | –150 |
a Cannot be measured.
We studied, therefore, [25] the isomerization of but-1-ene sorbed in CuZSM-5 containing both Cu+ ions and relatively strong Brønsted acid sites: Si–OH–Al formed by the hydrolysis of Cu2+. We compared the rate of isomerization of but-1-ene bonded to Cu+ ions (characterized by 1540 and 1570 cm–1 bands) and physisorbed but-1-ene (1640 cm–1 band). In both cases the migration of protons was necessary to produce carbenium ions and catalyze isomerization.
But-1-ene isomerization is accompanied by a decrease of the 1630 cm–1 band of C=C– vinyl group stretching and in the appearance of a much weaker 1645 cm–1 band of –C=C– stretching in vinylidene group in cis-but-2-ene (this vibration in trans-but-2-ene is IR inactive).
The spectra recorded upon the sorption of but-1-ene in CuZSM-5 at 210 K and upon the heating to 285 K are shown in Fig. 4. The spectrum of but-1-ene adsorbed at 210 K shows the bands of molecules: bonded to Cu+ ions (1540, 1570 cm–1), hydrogen bonded to hydroxyl groups (1630 cm–1) as well as of physisorbed molecules (1640 cm–1). Heating to 285 K resulted in a distinct decrease of both the 1630 and 1640 cm–1 band without any change in the 1540 and 1570 cm–1 bands of but-1-ene bonded to Cu+. This indicates that the isomerization of but-1-ene molecules bonded to Cu+ was much slower than for molecules hydrogen bonded and physisorbed. Comparing the isomerization rate of but-1-ene physisorbed and bonded to Cu+ shows that bonding but-1-ene with the Cu+ protected the molecule from the attack of proton, carbenium ions formation, and finally isomerization.

Izomerization of but-1-ene sorbed in CuZSM-5. But-1-ene sorbed at 210 K (a), and upon heating to 285 K (b).
It seems that the reason for the resistance of but-1-ene bonded to Cu+ to accept protons (even though it became negative because of π back donation) may be the fact that the carbon atom in –C=C fragment of but-1-ene strongly bonded to Cu+ has higher coordination number than that of a free or weakly bonded molecule so is less prone to accept protons from Brønsted sites.
The catalytic experiments [11] evidenced that CuMCM-41 was found to be more active in n-butenes isomerization. It is not excluded high activity of CuMCM-41 in skeletal isomerization of n-butenes may be due to a lower activation energy of another reaction steps, such as branching of secondary carbocation to tertiary one, or to abstraction of proton from tertiary carbocation what results in the formation of neutral isobutene molecule.
3.2.4 TPD-IR studies of butene desorption from CuX zeolite
TPD-IR studies of but-1-ene desorption [26] were undertaken to know if there are complexes Cu+-but-1-ene in CuX zeolite of various strength of Cu+ to butene bonding. The results shown in Figs. 1 and 2 suggest that the band around 1540 cm–1 may contain at least two submaxima. This is well seen in the case of ethene sorbed in CuY and barely seen for other alkenes. More information on the problem of heterogeneity of Cu+ sites was obtained in TPD-IR experiments of but-1-ene desorption from CuX.
In TPD-IR experiments, the sample is situated inside IR cell and IR spectra can be recorded. The process of adsorption can be followed by IR (the spectra at increasing loading can be analyzed), the status of adsorbed molecules and of zeolite modified by these molecules can be defined by IR spectroscopy, and the process of desorption can be followed both by IR spectroscopy and mass spectrometer. The scheme of the apparatus is shown in Fig. 5.
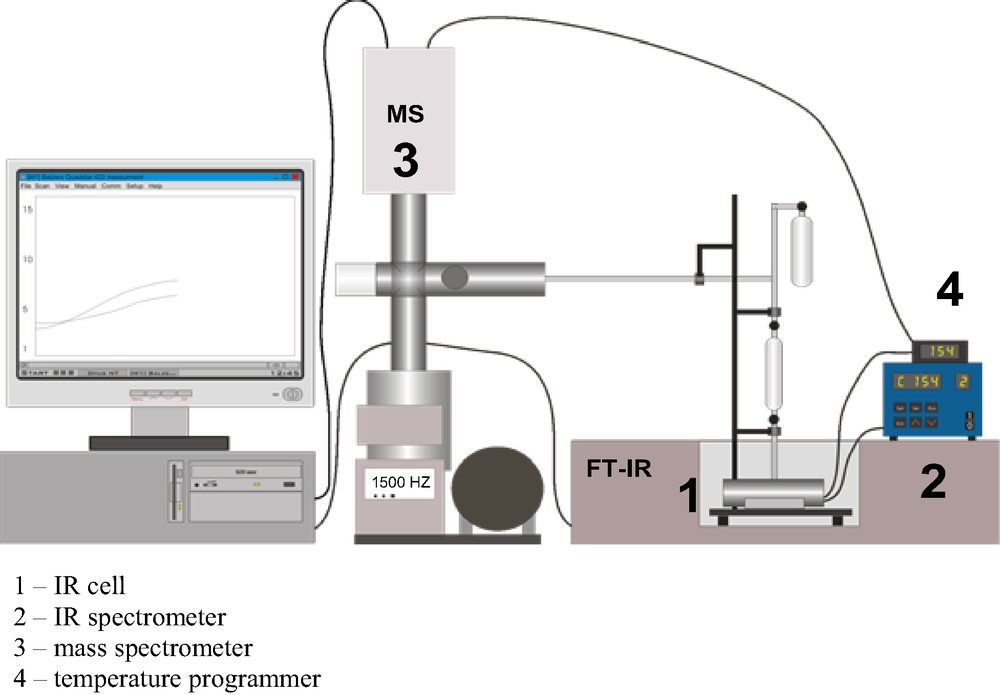
The scheme of apparatus used for TDP-IR experiments.
As mentioned above (Fig. 3A), the molecules of butene-1 adsorbed on CuX react with Cu+ ions in the first order (IR band at 1540 cm–1), and upon the saturation of Cu+ cations, they form hydrogen bonding with OH groups (band at 1630 cm–1) and the complex characterized by the band at 1570 cm–1. We studied thermoprogrammed desorption (TPD) of but-1-ene adsorbed on CuX, giving ca. 25% and 100% of Cu+ ions covered. In this second case (100% coverage) we made the affords to have only the complex characterized by the band 1540 cm–1, without butene-1 hydrogen bonded to OH groups nor butene-1 in the form of complex characterized by the 1570 cm–1 band.
At the beginning of TPD-IR experiment the portions of butene-1 were adsorbed until an appropriate coverage (estimated from the intensity of IR bands) was attained. These intensities were: 25% of the maximal intensity of 1540 cm–1 band and the maximal intensity of this band (but practically without the band at 1570 cm–1 and that of hydrogen bonded butene 1630 cm–1). The spectra of butene-1 adsorbed at coverage of 25% and 100% are shown in Fig. 6A, and corresponding them TPD diagrams in Fig. 6B. For low but-1-ene coverage, one TPD maximum around 470 K is present, whereas at full coverage a broad maximum with at least two submaxima: at 350 and 470 K was obtained.
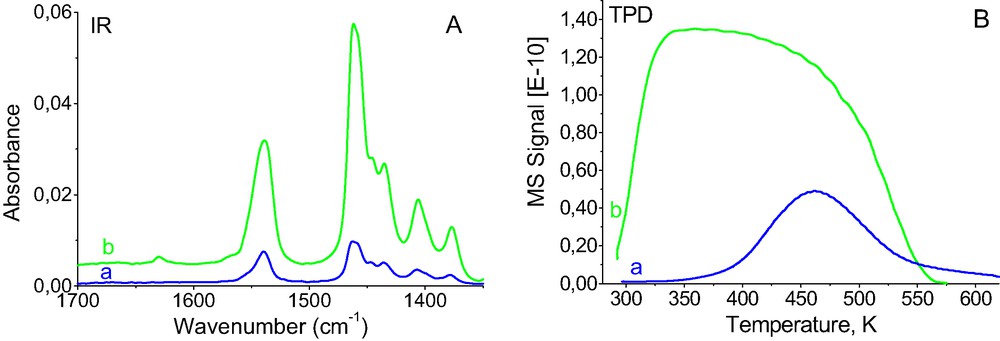
The results of TPD-IR experiments of but-1-ene desorption from CuX. (A) IR Spectra of but-1-ene adsorbed. (B) TPD Diagrams of but-1-ene desorption: (a) 20% of Cu+ covered by but-1-ene, (b) 100% of Cu+ covered by but-1-ene.
The result of TPD-IR experiments evidenced, therefore that at least two kinds of Cu+-but-1-ene complexes exist in CuX zeolite. Both of them are characterized by one IR band at 1540 cm–1 but they differ distinctly in the force bonding butene-1 with Cu+. The molecules of but-1-ene first adsorbed (at low coverage) form more favorable energetically complex in which the molecule is more strongly bonded to the cation. The analysis of IR spectra shown in Figs. 1, 2 and 6A suggests that 1540 cm–1 band is asymmetric and may contain some submaxima, but the frequencies of these possible submaxima are very close. Only the TPD-IR experiments evidenced that the butene-Cu+ complexes differing only a little in C=C stretching frequency can be bonded with considerable different force. This problem will be the subject of our further studies.
3.3 IR studies of acetylene sorption
Due to high symmetry of acetylene the vibrational spectrum of acetylene shows only five bands: three are active in Raman Spectroscopy and two in IR spectroscopy. IR active vibrations are: asymmetric ≡C–H stretching and asymmetric ≡C–H bending. The stretching of C≡C triple bond is IR inactive.
The interaction of acetylene with Cu+ sites in zeolite CuY was already followed by IR spectroscopy and by DFT [34]. In this research we followed by IR spectroscopy the interaction of acetylene with Cu+ in zeolites CuX, CuY and CuZSM-5.
The IR spectra of gaseous acetylene and acetylene adsorbed on SiO2, as well as sorbed in CuX are shown in Fig. 7. The C≡C stretching is IR inactive in free molecules but a weak band is seen upon the adsorption on SiO2 and hydrogen bonding with Si–OH–Al groups in HY zeolite, indicating that some molecules lost their symmetry when adsorbed. The small frequency shifts (Δν = 17 and 23 cm–1 for the molecules adsorbed on SiO2 and sorbed in HY) can be explained by hydrogen bonding formation with Si–OH and Si–OH–Al and by relatively weak π donation of electrons from π bonding orbitals of molecules to hydrogen atoms in hydroxyl groups.
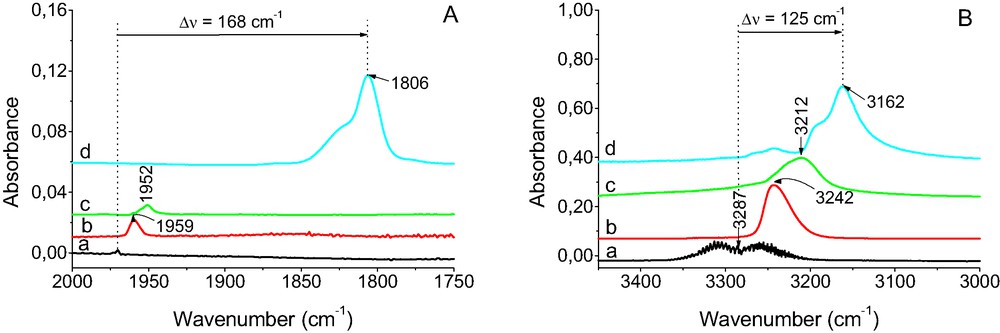
IR spectra of acetylene: (a) gaseous, (b) adsorbed on SiO2, (c) sorbed in zeolite HY, (d) sorbed in zeolite CuX.
In the case of sorption of acetylene in CuX zeolite a relatively intensive band at 1804 cm–1 of C≡C is present. The very high value of ΔνC≡C (168 cm–1) indicates very strong activation of C≡C bond by Cu+ ions in zeolites. This C≡C bond weakening was more distinct than the weakening of C=C bond in the case of alkene, e.g. for ethane ΔνC=C was only 78 cm–1.
It may be supposed that similarly as in the case of alkenes, π back donation of d electrons of Cu+ in zeolites to π* antibonding orbitals of acetylene takes place. The interaction of acetylene with Cu+ ions was also a subject of our quantum chemical DFT calculations and will be discussed in farther chapters.
Similarly as in the case of ethane, the stretching vibration of C≡C, which was IR inactive in free molecules, became IR active when acetylene interacts with Cu+.
According to the data shown in Fig. 7B, the band of ≡C–H asymmetric stretching vibration is also perturbed when acetylene interacts with Cu+. The value of frequency shift of ≡C–H stretching band Δν = 125 cm–1 of acetylene interacting with Cu+ is higher than for molecules interacting with Si–OH and Si–OH–Al groups (Δν = 45 and 72 cm–1, respectively). Similar phenomena were also observed in the case of alkenes (Figs. 1 and 2) but for acetylene the value of frequency shift of ≡C–H stretching band (Δν = 125 cm–1) is higher than for =C–H in alkenes (12–30 cm–1).
The spectra of acetylene sorbed in other Cu–zeolites: CuY and CuZSM-5 showed similar bands as in the case of CuX. In all the cases the band at ca. 1800 cm–1 is present, indicating that a strong activation of C≡C bond by Cu+ takes also place in other zeolites. Similar results were obtained by Hübner et al. [34].
The information on the heterogeneity of Cu+ sites in CuX zeolite interacting with acetylene was obtained in experiments in which the small doses of acetylene were sorbed at room temperature in CuX zeolite and in desorption experiments. The results of such experiments are shown in Fig. 8. The sorption of small amount of acetylene resulted in appearance of 1806 cm–1 C≡C band (Δν = 168 cm–1) and a second band at 1822 cm–1 (Δν = 152 cm–1) at higher acetylene loading. The desorption removed the species represented by 1822 cm–1 band in the first order (at lower temperatures) and those represented by 1806 cm–1 one in the second order (i.e. at higher temperatures).
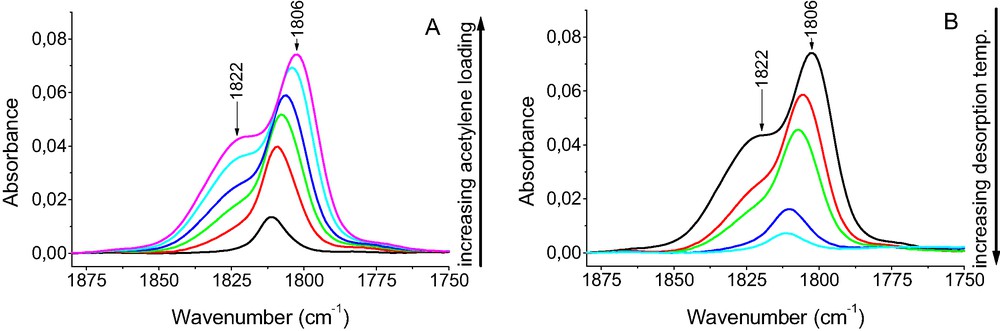
The spectra recorded upon the sorption of small doses of acetylene in CuX zeolite (A) and upon desorption at increasing temperatures (B).
Both adsorption and desorption experiments evidenced that at least two kinds of Cu+ sites exist in CuX zeolite, differing not only in the strength of acetylene bonding but also in the electrodonor properties. The sites of stronger electrodonor properties, i.e. of more pronounced π back donation power, activating C≡C bond stronger (νC≡C = 1806 cm–1) bound acetylene also stronger than Cu+ sites activating acetylene weaker (νC≡C = 1822 cm–1).
The results of our DFT quantum chemical calculations concerning the interaction of acetylene with Cu+ sites in zeolites will be presented in the farther chapter.
3.4 IR studies of benzene sorption
IR spectra of benzene shows one distinct band of ν19 C–C vibration of aromatic ring at 1480 cm–1. This band is well seen in the spectrum of benzene dissolved in CCl4 (Fig. 9). The interaction of benzene with Cu+ sites in CuZSM-5 results in a red shift of this band by 13 cm–1, indicating some weakening of C–C bond in aromatic ring.
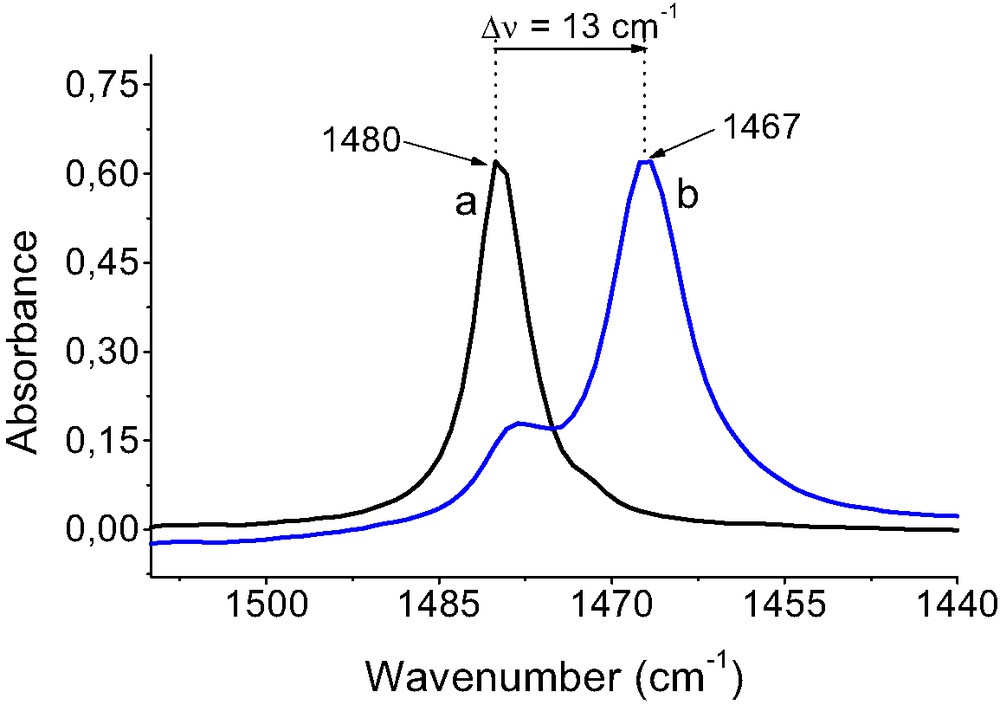
The spectra of benzene: (a) dissolved in CuCl4, (b) sorbed in CuZSM-5 zeolite.
The sorption of benzene in other Cu–zeolites (CuX and CuY) resulted in the appearance of the IR band at ca. 1470 cm–1 – similarly to that observed in the case of CuZSM-5 (Fig. 9). The results obtained for CuX are shown in Fig. 10. The spectra shown in Fig. 10A were recorded upon the sorption of small doses of benzene at room temperature. The bands at 1478 cm–1 of benzene bonded to Na+ ions still present in CuX zeolite and at 1468 cm–1 of benzene bonded to Cu+ ions appeared simultaneously and the ratio of their intensities did not vary distinctly with the loading. It indicates that benzene sorbed even at low loading reacted both with Cu+ and Na+ ions. The desorption experiments (Fig. 10A) showed that the band at 1478 cm–1 of benzene bonded to Na+ decreased first (at lower temperature) than that at 1468 cm–1 of benzene bonded to Cu+ ions. It indicates that the bonding of benzene with Cu+ ions is stronger than with Na+ ones. The fact, that at adsorption experiments the intensity ratio of 1468 and 1478 cm–1 bands does show only small variation with loading may be explained by assuming the benzene molecules react with Na+ and Cu+ sites without choosing more energetically favorable ones. Next the molecules bonded to less favorable Na+ desorb and readsorb on more favorable Cu+ ones. As desorption is activated process it is slow at relatively low (room) temperature.

The spectra recorded upon the sorption of small doses of benzene in CuX zeolite (A) and upon desorption at increasing temperatures (B).
We suppose that C–C bond weakening is the result of π back donation of d electrons of Cu+ to antibonding orbitals of benzene, similarly as observed in the case of alkenes and acetylene. According to the data given in Table 1 benzene is relatively good electron acceptor. The electron affinity value 1.3 eV is lower than for alkenes and acetylene: 1.4–1.5 eV. However, contrary to alkenes, LUMO orbital of benzene has only little antibonding (or even non-bonding) character. Therefore, the transfer of electrons from electrodonor Cu+ site to this orbital result in a very small weakening of C–C bond in aromatic ring. This explains a minute red shift of C–C band (13 cm–1 as seen in Fig. 9). It should be kept in mind that such a shift was 100–130 cm–1 in alkenes and 168 cm–1 in acetylene.
3.5 IR studies of acetone sorption
IR spectra of C=O group in acetone are shown in Fig. 11. Free acetone molecule (acetone dissolved in CCl4—Fig. 11, spectrum a) shows a distinct band at 1718 cm–1. The sorption of acetone in silicalite (spectrum b) resulted in appearance a new band at 1710 cm–1 of acetone hydrogen bonded to Si–OH groups present, to some extent, in silicalite. This last band shows a submaximum at 1695 cm–1. This shoulder (1695 cm–1 one) become the main band in the spectrum of acetone adsorbed on SiO2, in which the concentration of surface Si–OH groups is very high. The hydrogen bonding of Si–OH groups with acetone is also well seen in the region of O–H vibrations (spectra not shown). The sorption of acetone in HZSM-5 zeolite containing both Si–OH and acidic Si–OH–Al groups results in an appearance of similar bands as for SiO2: 1710 and 1695 cm–1, but the latter shows a submaximum at low frequency side (around 1685 cm–1), which may be assigned to the hydrogen bonding of acetone with acidic Si–OH–Al.

IR spectra of acetone: at solution in CCl4 (a), as well as adsorbed on silicalite (b), SiO2 (c), zeolite HZSM-5 (d) and zeolite CuZSM-5 (e).
The sorption of acetone in CuZSM-5 resulted in the appearance of all mentioned above bands: physisorbed acetone (1718 cm–1), acetone bonded to both Si–OH and Si–OH–Al groups (1710 and 1695–1685 cm–1), as well as a new maximum at 1679 cm–1 which is absent in zeolites without Cu. This band can be assigned to acetone bonded to Cu+.
The stability of acetone complexes bonded to OH groups and to Cu+ ions was studied in the experiments in which the small doses of acetone were sorbed in CuZSM-5, as well as in desorption experiments. The results are shown in Fig. 12. The sorption of small doses of acetone (Fig. 12A) resulted in the appearance of the bands of acetone bonded both to OH groups and to Cu+ ions. At high loading the band of physisorbed acetone 1722 cm–1 is present. The desorption at increasing temperature (Fig. 12B) resulted in the decrease of all the acetone bands but the bands of physisorbed acetone (1722 cm–1) and acetone bonded to OH groups 1710 and 1695 cm–1, whereas the band of acetone bonded to Cu+ (1679 cm–1) remains even at high desorption temperature. This results indicate that Cu+ sites bond acetone more strongly than hydroxyl groups.
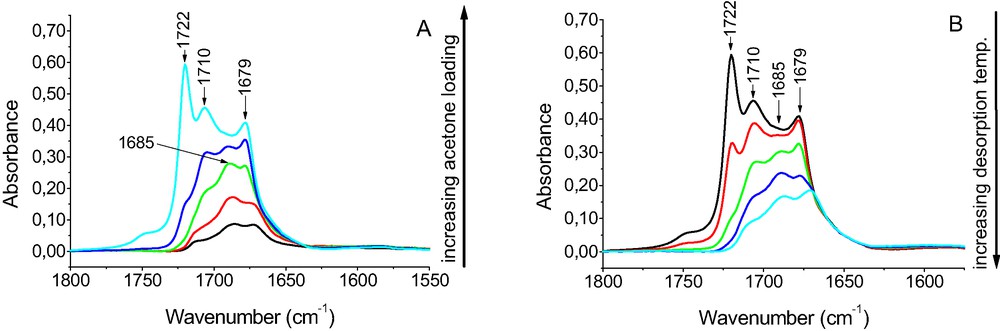
The spectra recorded upon the sorption of small doses of acetone in zeolite CuZSM-5 (A) and upon desorption at increasing temperatures (B).
The red shift of IR band of C=O interacting with Cu+ by 38 cm–1 indicates that the activation of C=O bond takes place. This effect is similar as that observed in the case of alkenes. Most probably the activation of C=O bond is realized similarly as for other molecules: alkenes, acetylene and benzene, which is π back donation of d electrons of Cu+ to antibonding π* orbitals of acetone.
According to the data given in Table 1, acetone is a good electron acceptor, its electron affinity value: 0.8 eV is the lowest of all organic molecules studied in this research.
3.6 IR studies of the effect molecules sorbed on the framework vibrations of CuZSM-5 zeolites
The IR studies of framework vibrations of zeolites may be an important source of information on the properties of cations and their interaction with adsorbed molecules. This method based on observation of the IR band in the ‘transmission window’ was first proposed by Sarkany [35–39] and then developed by other authors [40–43].
Generally, the introduction of cations into cation positions of MFI zeolites, as well as of other zeolites (e.g. ferrierites), resulted in some deformation of the oxygen ring and therefore in a shift of IR band of ring vibration from ca. 1020 cm–1 (position of unperturbed rings) to lower frequencies. A weak maximum or a shoulder between 930 and 980 cm–1 (in the so called ‘transmission window’) appeared, the position of which depended on the extent of deformation of the ring by cation and their intensity on the cations amount. The interaction of cations with adsorbed molecules such as CO, NO, NO2, NH3, H2O resulted in some cation withdrawal from the oxygen ring and in the ring relaxation. The IR band of ring vibration shifted to a higher frequency toward the position of unperturbed ring. Therefore, the observation of the band of oxygen ring vibration (ca. 930–980 cm–1) may be helpful in studies of the properties of cations in zeolites and their interaction with adsorbed molecules. This is especially helpful in the case of highly siliceous zeolites in which the properties of cations can not be studied by the XRD method.
The spectra of CuZSM-5 ‘in transmission window’ are shown Fig. 13. The spectra recorded at 170 K before and after the sorption of but-1-ene, ethene, benzene and acetone as well as different spectrum are given. A weak band at 960 cm–1 seen in the spectrum of activated CuZSM-5 can be interpreted as the vibration of oxygen rings deformed by Cu+. The effect of the adsorption of alkenes, benzene and acetone can be the best seen in different spectra. The band shift (Δν) was 22, 26, 26 and 36 cm–1 for benzene, acetone, ethene, and but-1-ene adsorbed, respectively. The comparing of these Δν values suggests that but-1-ene has the highest power to withdrew Cu+ cation from zeolitic oxygen ring and benzene is the less effective.
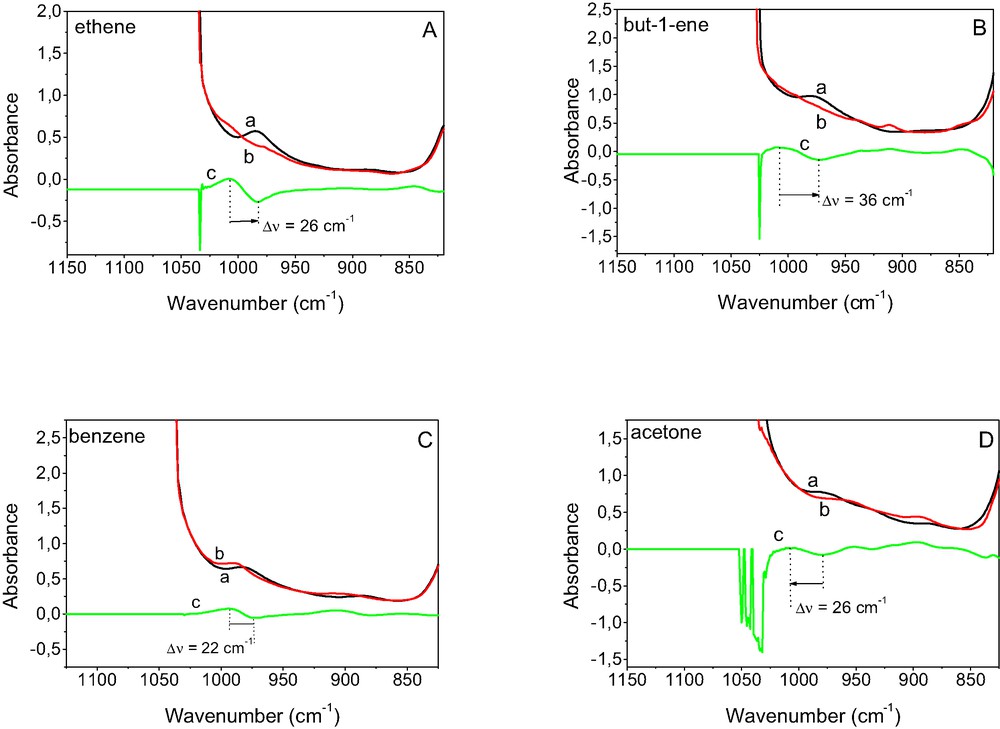
The IR spectra of CuZSM-5 recorded (at 170 K) at ‘transmission window’ upon the sorption of but-1-ene (A), ethane (B), benzene (C) and acetone (D). Spectra were recorded before (a), and after the sorption (b), as well as difference spectra c = b – a.
These Δν values were distinctly higher than for CO adsorbed (Δν = 7 cm–1 Ref. [34]), proving that relaxation of the oxygen ring by withdrawing Cu+ by alkenes was more pronounced than by CO. These results indicate that the interaction of Cu+ with organic molecules studied here was very strong, much stronger than with CO.
3.7 Quantum chemical calculation protocol and results concerning the interaction of alkenes and acetylene with Cu+ sites
As mentioned above, our calculations have been performed for a model of cationic copper site that was cut off the MFI structure taken from Accelryss databases included in the software. The basket (M7) model was composed of two fused 5T rings forming a 6T ring which simulated framework environment for copper cations in α position on main channel wall of MFI structure. This model has already been elaborated by us and studied in more detail previously [5–7]. The calculations evidenced, that copper cation (Cu+) was coordinated by four framework oxygen atoms with the following bond distances: 2.04, 2.05, 2.24, 2.43 Å. (Fig. 14).
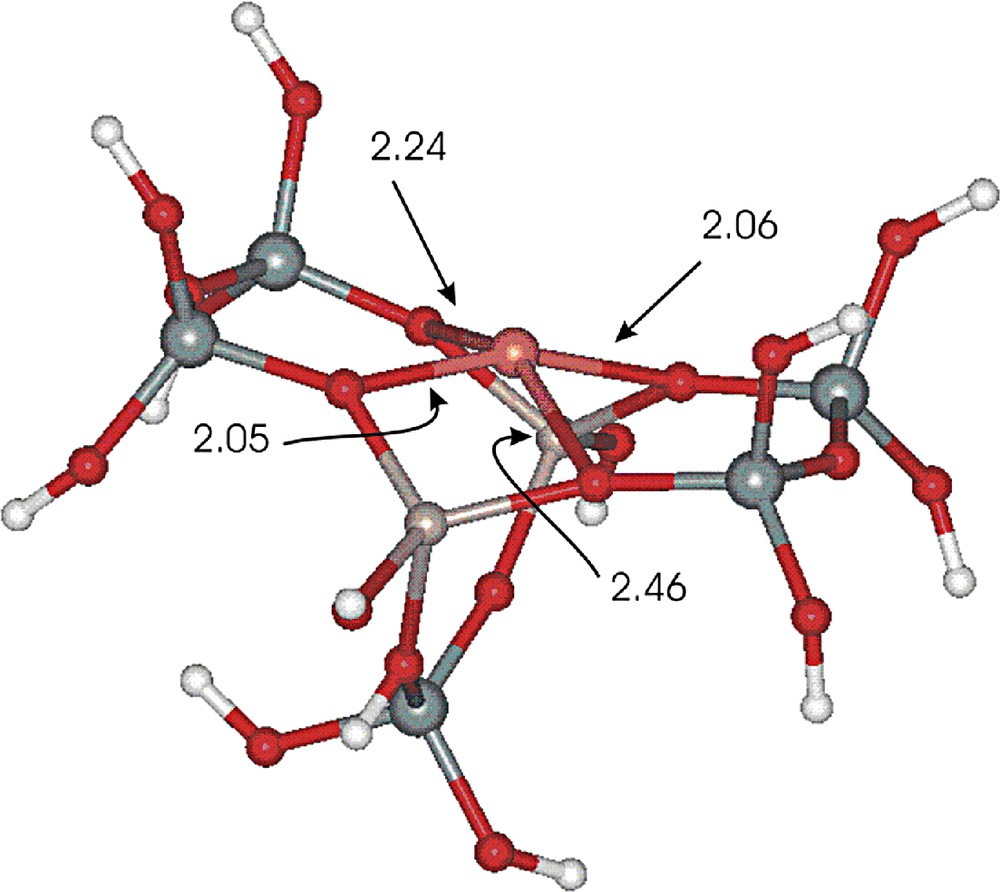
The model of copper cationic center in zeolite ZSM-5.
Molecules from the early alkene’s homologous series (ethene, propene, but-1-ene, cis-but-2-ene, trans-but-2-ene) and acetylene have been studied. The interaction of the same molecules with Cu+ ions in zeolites was followed by IR spectroscopy and has been discussed in previous chapters. Optimized geometry of the isolated molecules and their vibrational frequencies were used as a reference to describe changes imposed by the interaction with the Cu+ site (Table 2). As well calculated length of multiple bond in isolated hydrocarbon molecules as calculated frequencies matched reasonably well experimental expectations. Then each hydrocarbon molecule was put in the range of influence of the copper site. Such six composed systems have been subsequently optimized with preserved constraints for terminating hydrogens to simulate the influence of the remaining framework. All the molecules showed to be bound by the site as the result of optimization.
Length of double (triple in acetylene) bond RC=C (Å), calculated and experimental frequency of C=C (C≡C in acetylene) (cm–1) in isolated molecules
| Ethene | Propene | But-1-ene | Cis-but-2-ene | Trans-but-2-ene | Acetylene | |
| RC=C | 1.330 | 1.332 | 1.331 | 1.337 | 1.333 | 1.21 |
| 1658 | 1683 | 1688 | 1728 | 1714 | 2011 | |
| 1613 | 1646 | 1644 | 1663 | 1680 | 1950 |
In the first coordination sphere of Cu+ carbon atoms of double bond are equally distant from Cu+ (by ca. 2 Å). The cation looses two framework contacts and remaines equally bonded to two framework oxygen atoms (Table 3). Two oxygen atoms and two carbon atoms form approximately square planar coordination except for trans-but-2-ene, which is midway between square and tetragonal geometry. The closer to the square coordination the smaller the angle between the directions determined by two oxygen and two carbon atoms defined by the CCOO torsion angle (see Table 3). Thus this angle may be regarded as a measure of the out-of-plane distortion in copper coordination. Zero angle corresponds to square coordination while for tetragonal one it reaches 90°. Approximate square (ethene) and half-tetragonal (trans-but-2-ene) coordination is shown in Fig. 15a, b, respectively.
Geometry of the composed systems: average Cu–O and Cu–C distances, C=C bond length (Å), angle of distortion from square coordination (see text) of copper (deg)
| Ethane | Propene | But-1-ene | Cis-but-2-ene | Trans-but-2-ene | Acetylene | |
| Cu–O | 2.02 | 2.05 | 2.05 | 2.03 | 2.08 | 1.98 |
| Cu–C | 1.99 | 2.02 | 2.02 | 2.04 | 2.04 | 1.94 |
| C=C | 1.380 | 1.382 | 1.377 | 1.387 | 1.383 | 1.25 |
| Angle | 5 | 11 | 26 | 23 | 48 | 7 |
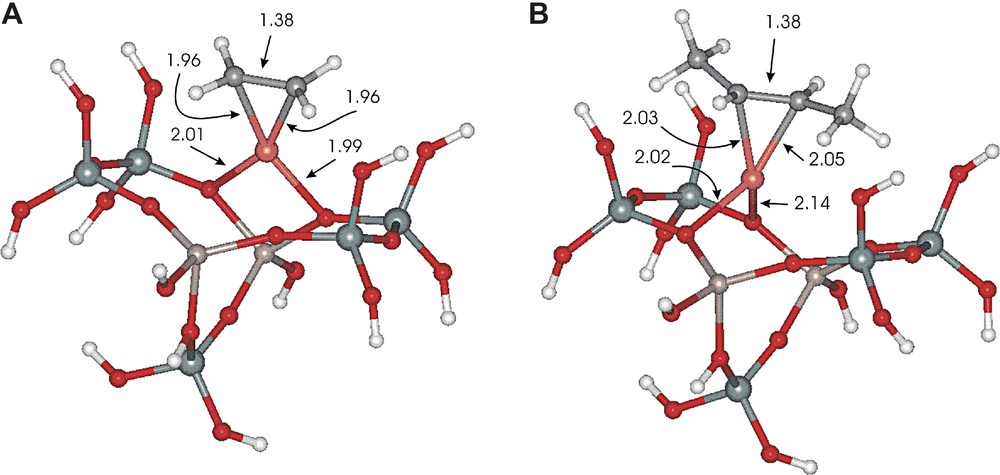
The copper cationic center interacting with ethene (A) and trans-but-2-ene (B).
All the studied molecules demonstrated significant interaction with the Cu+ site. It must be noted, however that apart from the interaction between a double bond and a cation, some hydrogen atoms of hydrocarbon molecule are close enough to form hydrogen bonds with framework oxygen atoms. These bonds may contribute partly to overall higher adsorption energy (ca. 200 kJ mol–1, see Table 4) than it is in the case of diatomic molecules like NO (ca. 100 kJ mol–1—Ref. [8]). This result has also been confirmed by IR experiments. The adsorption of but-1-ene after preadsorption of CO or NO leads to the exchange of previously adsorbed molecules, which formed quite stable complexes themselves: Cu+CO and Cu+(NO)2 [25].
Strong interaction between alkene molecules and a cationic site is concentrated in double bond region. The results of calculations indicate (Table 4) that in all cases the double bond becomes lengthened by ca. 0.05 Å and Mayer bond order for this bond decreases by ca. 0.6—the bond may be classified somewhat half-way between single and double bond. Calculated frequencies of the double C=C bond in adsorbed molecules are lower than these for free molecules by ca. 130 cm–1 (see Table 4). Changes in geometry, bond order and frequencies indicate that the bonds are strongly activated what nicely matches experiment. The change in frequency of C=C stretch could be the measure of the activation of the bond. The calculated and measured red shifts of IR C=C stretching bands agree well; the only exception is ethene where only qualitative agreement has been reached—possibly due to strong anharmonic contributions. The values of absorption coefficients of C=C band of alkenes have been calculated besides of stretching frequencies. They are zero for free molecules of ethene (D2h), trans-but-2-ene (C2h), and acetylene (D∞h). The interaction between these molecules and the Cu+ site breaks the symmetry, and therefore, the transitions attributed to C=C vibration become IR allowed. In spite of the fact that symmetrical molecules, e.g. ethane were adsorbed side-on, the vibration became active in IR due to the distortion of hydrogen atoms from previously planar or linear configuration in a free molecule. The increase in calculated and measured absorption coefficient correlates well with the out-of-plane distortion of the molecule measured by the sum of HCC angles in acetylene or HCCH dihedral angle in ethene and trans-but-2-ene (Table 5). Perturbation in geometry results in mixing of C=C and C–H vibrations and thus previously forbidden transition becomes allowed after adsorption of the molecule. It would never be the case if it were a diatomic homonuclear molecule adsorbed side-on. According to the data given in Table 5 the highest value of C=C extinction coefficient was obtained for acetylene and the lowest for trans-but-2-ene. This calculation result agrees with the experimental results shown in Figs. 1, 2 and 7. The C=C vibration is IR active in ethene and acetylene interacting with Cu+ in zeolites, but it remains IR inactive in trans-but-2-ene. It should be noted, that this would never occur for homonuclear diatomic molecule adsorbed side-on on adsorption site. In contrast, the N≡N vibration was found active in the case of N2 adsorbed on Cu+ in zeolites where the end-on adsorption probably occurs [44].
Symmetry distortion for ethene, trans-but-2-ene, and acetylene (deg); calculated absorption coefficient (cm μmol–1)
| Ethene | Trans-but-2-ene | Acetylene | |
| Angle | 13 | 11 | 34 |
| ɛ | 0.6 | 0.3 | 1.5 |
Moreover, as a rule calculated frequencies of either stretching or deformation vibrations engaging =C–H bond neighboring to double bond decrease. Contrary, unless hydrogen is bounded by hydrogen bond, the frequencies of ordinary H–C– vibrations in CH3 or CH2 groups more distant from the double bond change only a little in comparison with the frequencies calculated for free molecules. The same observation emerged also from our IR studies of alkenes sorption in zeolites (Figs. 1 and 2). This conclusion agrees with the assumption that alkene molecules interact with cationic site via double bond—the more distant the region of the molecule from the double bond the smaller are the changes.
The analysis of charge distribution gives deeper insight into the process of activation. As mentioned above, the calculated change of charge on copper cation from +1.0 for free catiaons to +0.32 after placing the cation in a zeolite framework suggests that the embedding may strongly influence its π donor properties. It shows also the function of a zeolite as an electron reservoir enhancing the ability of Cu+ to π back donation. However, the phenomenon of flowing electrons from the site to an interacting molecule may be also moderated by electron affinity of the molecule. As mentioned, the electron affinity reveals the ability of a molecule to be an acceptor of π back donation. Thus we have calculated electron affinities for a series of studied molecules as the difference between total energy of an anion and neutral molecule (see Tables 1 and 6). One can expect that the smaller total energy loss on electron accepting (the higher effective electron affinity) the more electrons may be gained by the molecule leading to its activation. Calculated energy loss for alkenes is ca. 1.4 eV, what is less than that for NO (0.5 eV) but more than that for CO or N2 (1.9 eV). Therefore, alkenes are more prone to π back donation than CO and N2 while NO is the best electron acceptor. The values of the electron affinity for alkenes suggest that alkenes could be activated by π-back donation while interacting with Cu+ site, which was already shown to have high ability to π back donation.
Electron affinity (EA) for free molecules (eV), change of charge on studied molecules QM and embedded copper cation in zeolites QCu after adsorption of hydrocarbons
| Ethene | Propene | But-1-ene | Cis-but-2-ene | Trans-but-2-ene | Acetylene | |
| EA | 1.7 | 1.5 | 1.4 | 1.7 | 1.5 | 1.5 |
| QM | –0.05 | –0.06 | –0.06 | –0.09 | –0.06 | +0.03 |
| QCu | +0.04 | +0.04 | +0.04 | +0.04 | +0.04 | +0.03 |
The extent of activation of the appropriate bond in a molecule after accepting the electron due to π back donation depends obviously on the character of the accepting molecular orbital. Thus the activation of alkenes and acetylene is significantly pronounced since their LUMOs are strongly antibonding.
Some additional information is brought about by the calculations for reference systems, which were free Na+ and Cu+ cations (i.e. not embedded in a zeolite framework). In these systems large charge transfer (0.25 ÷ 0.40 e) from the molecule to the center is registered. Although due to the large flow of electrons in that case alkene molecules become positive, the double bond is also weakened while an alkene molecule interacts with free cations. The calculated C=C frequencies for alkenes with free cation showed the red shift with respect to a hydrocarbon comparable to the shift calculated for alkene with Cu+ active site. IR experiments also evidenced that the interaction of but-1-ene with Na+ in zeolite shifts IR C=C bond by ca. 20–30 cm–1. This indicates that π-donation, obviously effective in this case must have smaller impact on the bond activation than π-backdonation (larger charge transfer needed for comparable effect). It follows also from the well-known principle of quantum chemistry that the bonding character of bonding orbitals is much less pronounced than the antibonding character of antibonding orbitals. Thus the analysis of charge distribution shows that in the case of free cations π-donation, even if effective, must be much smaller than in the case of cation embedded in a zeolite framework where the activation is very strong. Therefore, the function of a zeolite acting as a reservoir of electrons which strongly enhances the ability of Cu+ cation to π-backdonation seems to be clearly documented.
The difference in the effects of the interaction between alkene molecules with the free or embedded cation consists in much more significant π-back donation from the cation to the molecules when surrounding of zeolite framework is present. Initially neutral molecules become negatively charged and positive Cu+ cation becomes even more positive (Table 6). These changes support more general observation that global flow of electrons takes place from the positive site to the adsorbed molecule and that copper cation serves as an electron transmitter.
On the basis of preceding discussion one can easily understand and rationalize the fact that zeolites serve as an excellent host for late transition metal cations, especially the copper, forming powerful catalytic active sites. In particular copper cationic sites in zeolites having great ability to strongly activate the C=C bond could be potential catalysts in reaction with alkenes as a substrate, e.g. in fine chemistry reaction.
4 Conclusions
1. The interaction of alkenes: ethene, propene, but-1-ene and cis-but-2-ene with Cu+ cations in zeolites resulted in a strong activation of the C=C bond. The double bond stretching band shifted to lower frequencies by 78–115 cm–1, indicating a distinct C=C bond weakening. The interaction of the C=C group with Cu+ cation also resulted in a shift of IR bands with stretching, deformation and bending of C–H bonds in =CH2 and =C–H groups in the vicinity of double bond.
2. The interaction of acetylene with Cu+ ions results also in a strong activation of triple C≡C bond and in a distinct bond weakening. The triple bond stretching band shifts by 168 cm–1 (more than in the case of double bond in alkenes). The ≡C–H stretching band does also shift by 125 cm–1. This effect is also distinctly stronger than in the case of alkenes.
3. In the case of ethene and acetylene the stretching of the C=C and C≡C bonds which were IR inactive in free molecules became IR active when interacting with Cu+, indicating the loss of symmetry. By contrast, the C=C stretching in trans-but-2-ene was still IR inactive when trans-but-2-ene interacted with Cu+.
4. At high loading some Cu+ ions were able to bond two alkene molecules.
5. But-1-ene molecules bonded to Cu+ ions were less prone to accepting protons from Brønsted acid sites and to isomerization. It is possible that the reason is a higher coordination number of carbon atom bonded to Cu+ than in a free molecule.
6. Cu+ ions in zeolites were able to activate aromatic C–C bond in benzene. The red band shift was 13 cm–1, i.e. much less than in the case of alkenes and acetylene.
7. The activation of C=O bond in acetone by Cu+ in zeolits results also in the bond weakening. The red band shift was 38 cm–1.
8. Molecular modeling by DFT performed for the model of the copper cationic centers and hydrocarbon molecules confirm strong adsorption of ethene, propene, butenes, and acetylene.
9. Calculated properties such as vibrotional frequencies of the adsorbed molecules match well experimental trends.
10. The analysis of charge distribution reveals the function of zeolite as a host for transition metal cationic centers which act as electron transmitters. Flow of electrons and subsequent activation can be explained on the basis of electronic structure of the molecules, their electron affinity and symmetry as well as the ability of the site to π-backdonation.
Acknowledgments
This study was sponsored by Polish Ministry of Scientific Research and Informational Technology (grant No. 4 T09A 184 24).