

1 Introduction
The synthesis of π-conjugated oligomers has attracted considerable attention because of their use as models for precursors of conductive polymers and in technological applications, such as field-effect transistors or organic light-emitting diodes [1]. The diversification of their optical and electronic properties is a key issue for manifold applications of these materials [1e,1h]. This aim can be achieved by the incorporation of various heterocyclopentadienes (thiophene, pyrrole, silole…) into the backbone of π-conjugated systems, since the electronic properties of these building blocks are highly dependant on the nature of the heteroatom [1a–b,1h,2]. In this context, the phosphole ring is an attractive synthon since it displays electronic properties that are markedly different from those of the widely used highly aromatic thiophene and pyrrole rings [3]. The phosphorus atom does not readily form s–p hybrids and mainly employs p-electrons for bonding. This ‘nert-pair effect’ implies a pyramidal geometry of the tricoordinate phosphorus atom with the lone pair having a pronounced s-character. These geometric and electronic features prevent efficient interaction of the phosphorus lone pair with the endocyclic dienic system [3]. In fact, the delocalisation within the phosphole ring arises from a hyperconjugation involving the exocyclic P–R σ-bond and the π-system of the dienic moiety [4]. Thus, phospholes possess (i) a weak aromatic character, which should favour delocalisation of the π-system, and (ii) a reactive heteroatom, which potentially offers the possibility of tuning the HOMO and the LUMO levels via chemical modification such as coordination to metal ions. These unique properties, that set phosphole apart from thiophene and pyrrole, prompted us to prepare and to study the physical properties of π-conjugated materials containing phosphole rings. In this account, we describe the synthesis and properties of 2,5-di(heteroaryl)phospholes and we illustrate that their ligand behaviour opens the route to novel architectures exhibiting interesting optical and optoelectronic properties.
2 Co-oligomers containing phosphole ring
2.1 Symmetrical phosphole-derivatives
2.1.1 Synthesis
2,5-Di(heteroaryl)phospholes 3 (Scheme 1) were prepared via the ‘Fagan–Nugent method’, a general and efficient organometallic route to phosphole moieties [5]. The intramolecular oxidative coupling of functionalised 1,7-diynes 1 (Scheme 1), possessing a (CH2)4 spacer in order to obtain the desired 2,5-substitution pattern, with 'zirconocene' provides the corresponding zirconacyclopentadienes 2 [6]. These organometallic intermediates react with dihalogenophosphines to give the corresponding phospholes 3 in medium to good yields. It is noteworthy that the stability of these phospholes is intimately related to the nature of the P-substituent. 1-Phenylphospholes 3a, b can be isolated as air-stable solids following flash column chromatography on basic alumina. In contrast, 1-alkylphosphole 3c and 1-aminophosphole 3d are extremely air and moisture sensitive compounds [6b].
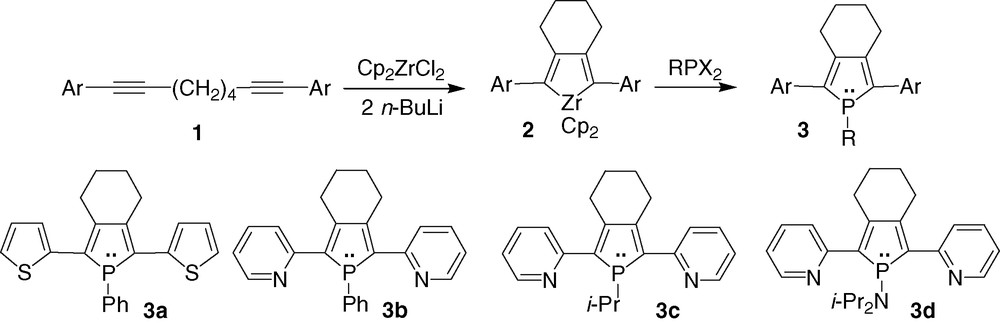
σ3,λ3-Phospholes 3a [6a] and 3b [7] (Scheme 1) bearing electron-rich and electron-deficient substituents, respectively, were characterised by X-ray diffraction studies. In spite of the different electronic nature of the two 2,5-substituents, compounds 3a, b share some important structural features in the solid state. Their molecular crystal structures reveal that the three heterocycles are almost coplanar while the phosphorus atom's environment is strongly pyramidalised [6,7]. Moreover, it is noteworthy that the carbon–carbon lengths between the rings are in the range expected for Csp2–Csp2 bonds. These solid state data indicate that an efficient delocalisation of the π-system takes place over the three heterocycles in oligomers 3a, b, as supported by theoretical studies [6a,8].
2.1.2 Physical properties
The presence of an extended π-conjugated system in phospholes 3a–d was confirmed by the observation of a broad absorption in their UV–vis spectrum. The energy of these absorptions, attributed to π–π* transitions [8], depends dramatically on the nature of the 2,5-substituants of the phosphole ring (Table 1) [6,7]. The values of λmax and the optical end absorption λonset (the solution optical ‘HOMO–LUMO’ gap) [1e], become red-shifted on replacing the 2-pyridyl groups (λmax = 390 nm) by 2-thienyl rings (λmax = 412 nm). Recent theoretical studies showed that this bathochromic shift is due to a better interaction between the HOMO of phosphole with the HOMO of thiophene, compared to that of pyridine [6b,8]. It is worth to note that the value of λmax recorded for 3a (412 nm) [6a,b] is considerably more red-shifted than that of the related tert-thiophene (355 nm) [9]. This observation is in agreement with theoretical studies predicting that heterocyclopentadienes with a low aromatic character are optimal building blocks for the synthesis of extended π-conjugated systems with low HOMO–LUMO gap [1h,6d].
Photophysical and electrochemical data in THF solutions at room temperature a and Td5 for compound 3a–9a and 3b
| λmax (nm), ɛ (mol–1 l cm–1) a | λem (nm) a | Φ (%) b | Epa (V) d | Epc (V) d | Td5 (°C) e | |
| 3a | 412 (8500) | 501 | 5 | +0.40 | – | 205 |
| 3b | 390 (10,500) | 463 | 1.1 | +0.83 | –2.45 | 210 |
| 3c | 371 (12,600) | 458 | – | +0.79 | –2.67 | 198 |
| 4a | 432 (9500) | 548 | 4.6 c | +0.68 | –1.95 | 253 |
| 4b | 364 (8700) | 470 | < 0.2 | +1.43 | –1.88 | 145 |
| 5a | 434 (9300) | 556 | 0.7 c | +0.63 | –2.03 | 214 |
| 6a | 442 (8300) | 593 | 0.8 c | +0.92 | –1.66 | 196 |
| 6b | 394 (9600) | 510 | 6.8 | > + 1.80 | –1.19 | 148 |
| 7a | 408 (10,900) | 506 | 1.3 c | +0.70 | –2.20 | 186 |
| 7b | 373 (10,230) | 465 | < 0.2 | +0.51 | –2.09 | 204 |
| 8a | 428 (15,100) | 544 | 14 c | +0.82 | –1.75 | 218 |
a Absorption and emission maxima, ± 3 nm.
b Fluorescence quantum yields determined relatively to quinine sulphate unless otherwise stated, ± 15%.
c Relative to fluorescein ± 15%.
d All potentials were obtained during cyclic voltammetric investigations in 0.2 M Bu4PF6 in CH2Cl2. Platinum electrode diameter 1 mm, sweep rate : 200 mV s–1. All reported potentials are referenced to the reversible formal potential of the ferrocene/ferrocenium couple.
e Thermal decomposition estimated by thermogravimetric analysis under N2, 5% weight loss.
Phospholes 3a–d are also fluorophores and their λem (Table 1) depend on the nature of the 2,5-substituents. A blue emission is observed for di(2-pyridyl)phosphole 3b (λem = 463 nm) whereas the emission of di(2-thienyl)phosphole 3a is red-shifted (Δλem = 35 nm). Cyclic voltammetry (CV) performed at 200 mV s–1 revealed that the redox processes observed for all the σ3-phospholes 3 are irreversible and that their redox properties are related to the electronic properties of the phosphole substituents. For example, derivative 3a featuring electron-rich thienyl substituents is more easily oxidised than compound 3b which possesses electron-deficient pyridyl substituents (Table 1) [6b].
In conclusion, co-oligomers with a phosphole core exhibit very interesting properties. They are air-stable providing that the P-substituent is a phenyl group, they exhibit low optical gap and their electronic properties can be tuned by varying the nature of the 2,5-substituants (2-thienyl or 2-pyridyl) of the phosphole ring.
2.1.3 Chemical modifications of the phosphole rings
2.1.3.1 Oxidation of the P-centre
In contrast with the more common group 14 and 15 heterocyclopentadienes (siloles, pyrroles), phospholes possess a reactive heteroatom offering the opportunity to prepare a family of π-conjugated systems starting from a single P-containing chromophore. The symmetrical phospholes 3a, b have been the subject of very simple organic reactions based on the nucleophilic behaviour of the σ3,λ3-phosphorus atom (Scheme 2) [6,7]. Derivative 3a, b have been treated with elemental sulphur, bis(trimethylsilyl)peroxide and methyl trifluorosulphonate affording σ4,λ4-phospholes 4a, b, 5a and 6a, b, respectively, in high yields [6a,b,7].
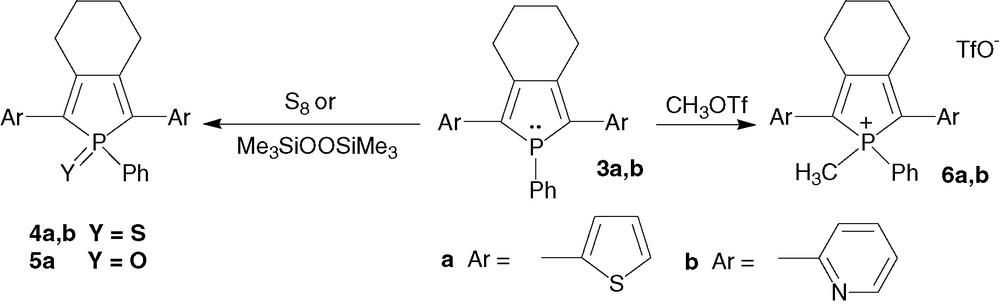
These chemical modifications allow to fine tune the electronic density of the P-atom and have a profound impact on the optical and electrochemical properties of these P-based oligomers. For both series, the gradual increase of the electron deficiency of the P-atom induces a bathochromic shift of both the λmax and the λem (3a/4a/6a, 3b/4b/6b, Table 1), an increase of the first oxidation and reduction potentials [6b]. The highest bathochromic shift following modification of the environment at phosphorus, was observed for the 2,5-dithienylphosphole derivatives (Table 1). Of particular interest, the thermal stability of oxo- and thiooxo-phospholes is higher than that of their precursors 3a, b. These results illustrate the contribution of phosphorus chemistry to tune the properties of π-conjugated systems.
2.1.3.2 Coordination chemistry of phosphole-based co-oligomers
Compared to organic molecules, metal complexes offer a larger variety of molecular structures, the possibility of high environmental stability and a diversity of electronic properties [10]. Since it is well-known that phospholes behave as classical two-electron donor tertiary phosphanes towards transition metals [11], we have explored the coordination behaviour of phosphole oligomers 3a, b with the aim to obtain complexes exhibiting interesting photophysical properties and original structures.
Tungsten(0) 7a, b and gold(I) 8a complexes are readily obtained as air-stable derivatives (Scheme 3). X-ray diffraction studies showed that their metric data (C–C inter-ring distances, P–C bond lengths…) are similar to those of the free ligands 3a, b [6a,7] and that the three heterocycles remain almost coplanar in the coordination sphere of the metal ions. These solid-state data indicate that the delocalisation of the π-system in complexes 7a, b and 8a still takes place over the three heterocycles. The electronic properties of these complexes depend on the nature of the metal ion. While the tungsten(0) complex's 7a photophysical properties are similar to those of the ligand 3a, the gold(I) complex 8a exhibited a red shift of both the λmax and the λem compared to 3a (Table 1).
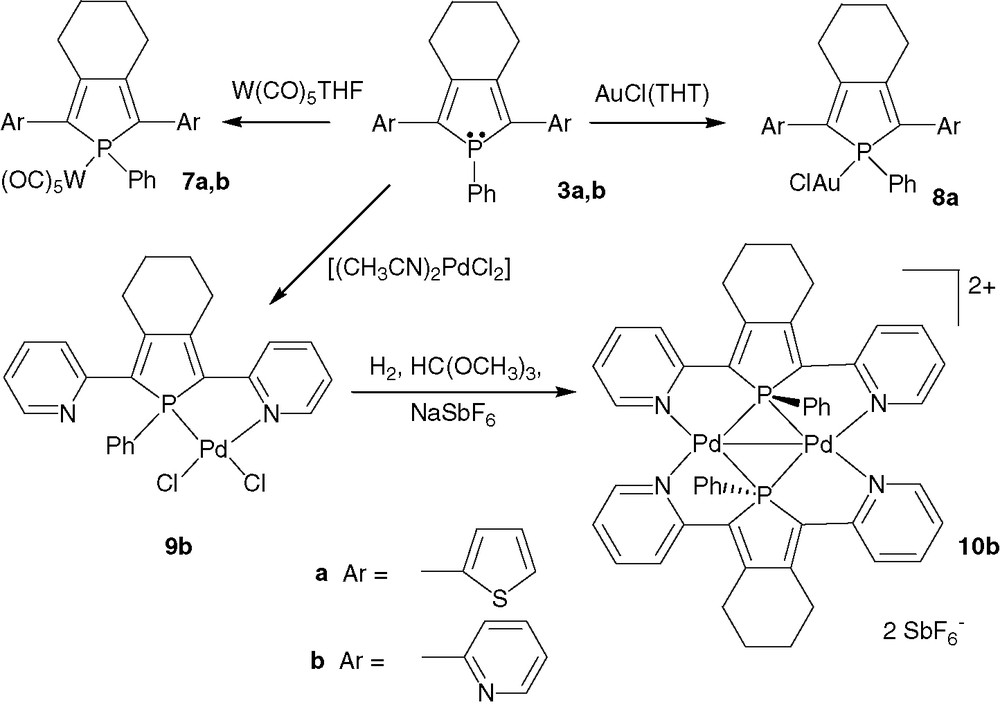
These mononuclear complexes played a great part in the development of phosphole-based derivatives in the field of OLED's. The free ligand 3a is not thermally stable enough to be sublimed and thus cannot be used as emissive material for OLEDs. In contrast, the gold complex 8a can be deposed on tin indium oxide by sublimation under high vacuum [12]. Single layer devices integrating 8a exhibited EL emission covering the 480–800 nm domain [12]; the low energy emissions, which are not observed for OLEDs prepared with thioxophosphole 4a, are very probably due to aurophilic interactions in the solid state [13]. This result clearly shows that the potential of phosphole-based oligomers for optoelectronic applications is considerably increased by the possibility to prepare coordination complexes.
2,5-Di(2-pyridyl)phosphole 3b can also act as 1,4-P,N chelate combining soft sp3 phosphorus and hard sp2 nitrogen centres [14] as illustrated by the synthesis of Pd(II) complex 9b. Under reducing conditions in presence of NaSbF6, 9b cleanly afforded complex 10b in which two ligands 3b act as six electrons μ-1χN:1,2χP:2χN donors toward a Pd(I)–Pd(I) core [15]. In this derivative, the P and N donors are in a mutual trans-configuration, in accordance with the anti-symbiotic effect (trans-influence) [16]. The most striking feature of complex 10b is that the two P-atoms adopt a symmetrically μ2-bridging coordination mode, a very rare coordination mode for phosphanes [17,18]. The UV–vis spectra of complex 10b exhibits several maxima between 300 and 500 nm due to σ(Pd–Pd) → σ*(Pd–Pd) transition and charge transfer from the metal or the phosphorus-metal fragments to the pyridine ligands [18]. The possibility for phosphole rings to act as symmetrically bridging ligands clearly opens new perspectives for the construction of original π-conjugated materials.
2.2 Unsymmetrical 2-(2-pyridyl)phospholes
The previous results prompted us to investigate ahead the coordination chemistry of 2-(2-pyridyl)phospholes with the aim to use the heteroditopic nature of these ligands to organise π-systems in the coordination sphere of transition metal and also to prepare polymetallic species.
2.2.1 Synthesis of 2-(2-pyridyl)phosphole ligands
Unsymmetrical 2-(2-pyridyl)phospholes 3e–h have been prepared in medium to good yields according to the Fagan–Nugent route starting from unsymmetrical diynes (Scheme 4) [5,15,19].
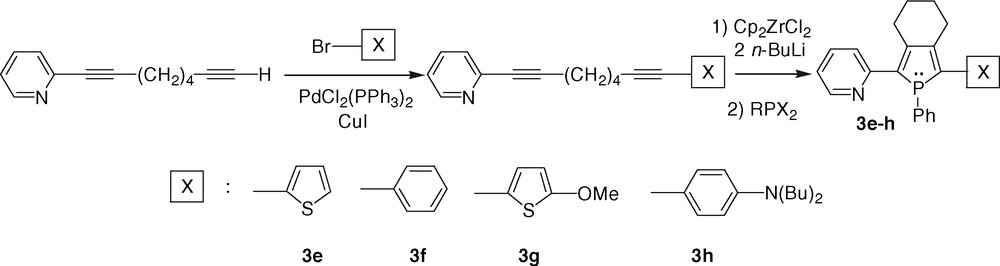
Theoretical studies have suggested that the weakly aromatic phosphole P-heterocycle is a potential interesting π-bridge for the engineering of dipolar donor–π-acceptor NLO-phores [20]. However, dipolar phospholes 3g, h (Scheme 4) exhibit rather low β values (ca. 30 × 10–30 esu), probably due to the weak accepting properties of the pyridine moieties [21].
2.2.2 Coordination chemistry
2.2.2.1 Palladium complexes
The coordination chemistry of 2-(2-pyridyl)phosphole ligands was investigated toward Pd(II) centres with the aim to gain more insight into their coordination behaviour. Thus, the reaction of 3e, f with [(cod)PdMeCl] (cod = 1,5-cyclo-1,5-octadiene) afford the corresponding air-stable complexes 4e, f as single diastereoisomer in excellent yields (Scheme 5) [14]. X-ray diffraction studies performed on complex 4f revealed a cis-arrangement of the phosphorous atom and the methyl group, as a consequence of the classical trans effect occurring in the coordination sphere of square planar d8 metal ions [16]. It is noteworthy that the cationic version of complex 4f turned out to be an efficient catalyst for carbon monoxide–olefin copolymerisation [14].
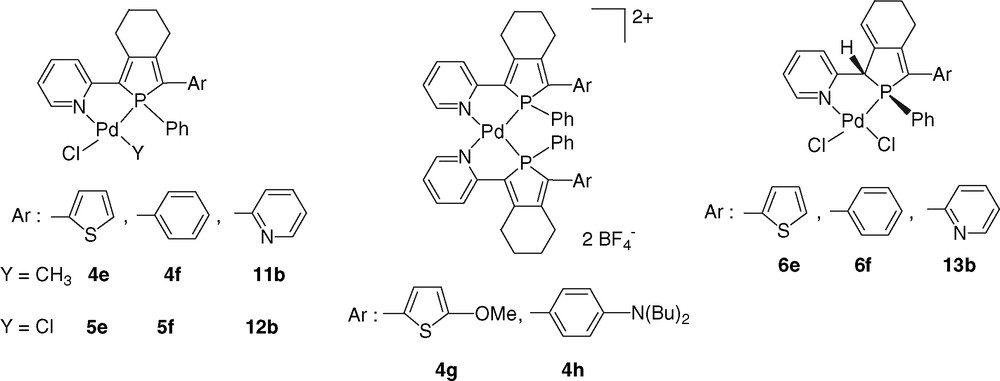
As already mentioned, phosphole-based dipoles 3g, h are not efficient NLO-phores. However, their potential in ONL is considerably increased by their coordination behaviour toward Pd(II) centres. The reaction of two equivalents of 2-(2-pyridyl)phospholes 3g, h with [Pd(CH3CN)4][BF4]2 afforded complexes 4g, h as single diastereisomers (Scheme 5) [19], with the P and N donor in a mutual trans-arrangement. Clearly, the trans-influence can overcome the natural tendency of 1D-dipolar chromophores to adopt an anti-parallel alignment. In other words, the square–planar metal centre acts as a template imposing a noncentrosymmetric assembly of two identical 1D-chromophores 3g, h. Furthermore, the metal plays a puzzling role since a considerable enhancement of the NLO-activities is observed upon complexation. For example, complexes 4h exhibit fairly high nonlinear optical activities with β values reaching 180 × 10–30 esu, a value which is much higher than the sum over the contribution of two sub-chromophores 3h. In a first approach, this effect could be related to an increase of the acceptor character of the pyridine groups [21]. However, it is very likely that the origin of this large β enhancement is due to metal-to-ligand (MLCT) or ligand-to-metal-to-ligand charge transfers (LMLCT) evidenced by UV–vis spectroscopy [19].
It is interesting to note that X-ray diffraction studies of complexes 4f and 5e revealed a significant degree of ring strain imposed by the formation of the five-membered metallacycle [14,22b]. This ring strain permits base-catalysed stereospecific [1,3]-H migration to afford complexes 6e, f and 13b containing 2-phospholene ligands [22]. X-ray diffraction studies showed that the H-atom linked to the C(1)-centre and the P-substituent adopt a mutually cis-configuration. This isomerisation process of coordinated phospholes into 2-phospholenes can be a serious drawback since the extended π-conjugated system of phosphole-based ligands collapses in the corresponding 2-phospholene derivatives.
2.2.2.2 Ruthenium complexes
A great attention has been paid to the synthesis and photophysical properties of dinuclear ruthenium complexes containing bridging π-conjugated ligands [23]. The design of the bridging ligands allows tuning of the electronic properties of these dyads and has led to important progress in the field of photoinduced electron or energy transfer processes. These properties prompted us to investigate a new family of derivatives possessing two terminal (2-pyridyl)phosphole moieties joined by a 2,5-thienyl spacer as potential bridging ligands to build bimetallic ruthenium complexes.
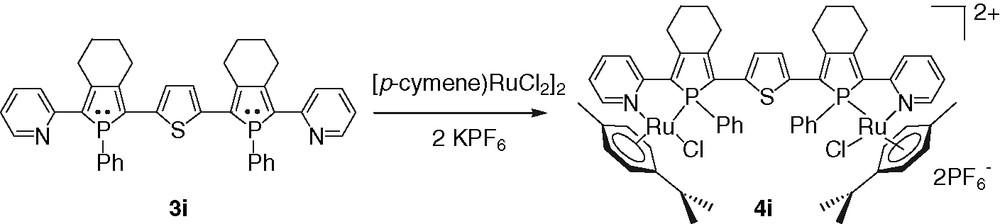
The target complex 4i was obtained by addition of [(p-cymene)RuCl2]2 and 2 equiv of KPF6 to the ligand 3i (Scheme 6) [24]. This coordination is diastereoselective, each diastereoisomer of 3i giving one diastereoisomer of 4i. The UV–vis spectrum of 3i exhibits an absorption maximum in the visible region at 482 nm attributed to π–π* transitions of the π-conjugated system [24]. This value is considerably red-shifted (Δλmax = 84 nm) compared to that of 3e. The coordination of the 2-pyridylphosphole moieties of 3i has almost no influence on the λmax. This is probably due to the symmetrical structure of 3i which prevents efficient ILCT. It is noteworthy that this broad ligand-centered transition overlaps the possible metal-to-ligand-charge-transfer transition arising from Ru(II) to π*-orbital of the coordinated 2-pyridylphosphole moiety.
3 Conclusion
This account illustrates the specific appealing advantages offered by the phosphole ring compared to its widely used sulphur or nitrogen analogues. First, thanks to their low aromatic character, phospholes are excellent building blocks for the tailoring of oligomers exhibiting low HOMO–LUMO gaps. Second, the possibility of operating chemical modification on the λ3-P centres gives a unique way to modulate the physical properties of these P chromophores. In this approach, coordination chemistry provided a wide range of original complexes based on monodentate phosphole-based co-oligomers, bidentate 2-(2-pyridyl)phosphole or tridentate bis[2,5-(2-pyridyl)]phosphole derivatives. The specific properties of these complexes (high thermal stability, predictable geometry, control of the coordination sphere…) considerably extent the potential of phosphole-based π-conjugated systems, as illustrated by their use in the field of nonlinear optics or electroluminescent diodes.