

1 Introduction
Over the last decade, electron-rich acetylides complexes stable under several redox states [1–4] emerged as promising building blocks for the realisation of various molecular devices [5–10]. With the objective of better understanding the electronic properties of organoiron representatives bearing the redox-active ‘(η2-dppe)(η5-C5Me5)Fe’ fragment (dppe = bis-1,2-(diphenylphosphino)ethane), [7,8] we have initiated an extensive study of series of Fe(II) and Fe(III) aryl acetylides possessing various X substituents in para position on the aryl ring [11–14]. In the course of these investigations, several attempts to grow crystals of the [(η2-dppe)(η5-C5Me5)FeC≡C–1,4-(C6H4)NMe2][PF6] radical cation (1+PF6–) actually resulted in the characterisation of the known iron(II) [(η2-dppe)(η5-C5Me5)FeCO][PF6] carbonyl adduct (2+PF6–), originally reported by Catheline and Astruc [15] (Fig. 1). We now report here the solid-state structure(s) of this unexpected product, as well as our investigations leading to a mechanistic proposal rationalising its formation from 1+PF6–.
Structures of 1+ and 2+.
2 Results
2.1 Crystal structures of 2+
Upon recrystallisation of the [(η2-dppe)(η5-C5Me5)FeC≡C(C6H4)NMe2][PF6] iron(III) complex (1+PF6–), large ruby-red cubic-shaped crystals corresponding to the cationic iron(II) carbonyl adduct 2+PF6–(A) rather than to the expected complex 1+PF6– were obtained after several weeks (Figs. 2 and 3a). In the hope of identifying the desired Fe(III) complex 1+, the isolated crystalline material was closely inspected and another type of crystal could be identified as pale-orange plates present in small amounts among the red crystals. These proved to constitute another structural variation (B) of 2+PF6– (Fig. 3b). Thus, in spite of our efforts, 1+PF6– was not identified among the crystalline material isolated. Finally, in order to dispose of crystals of 2+ with another counter ion for comparison purposes, we have crystallised an authentic sample of 2+BF4– using the same solvents (Fig. 3c).
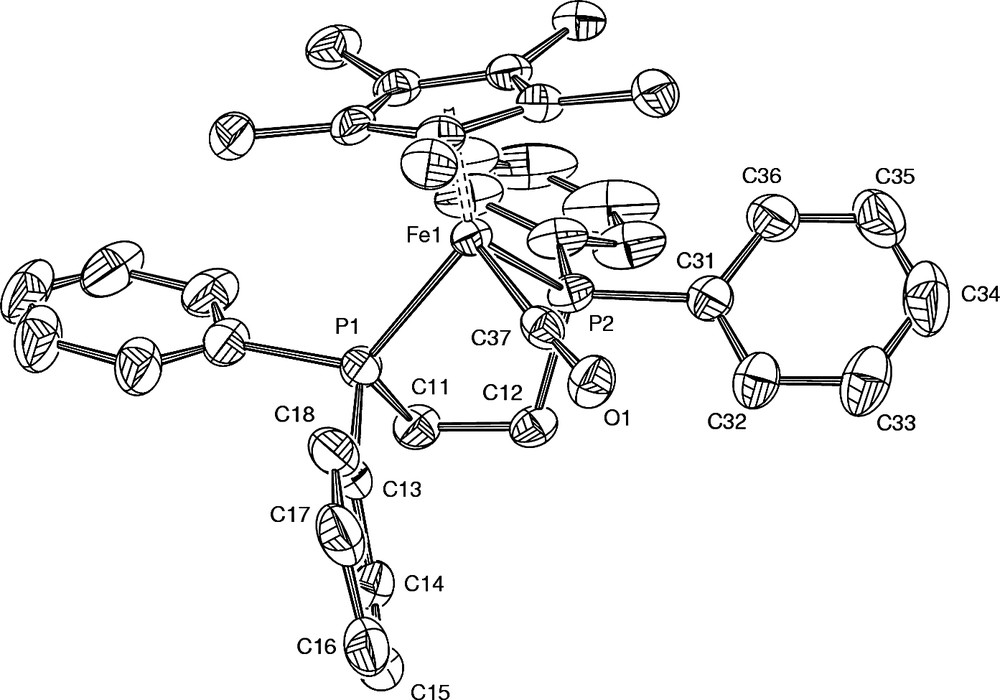
ORTEP drawing and labelling scheme for selected atoms of one cation of the asymmetric unit in 2+PF6– (polymorph A).
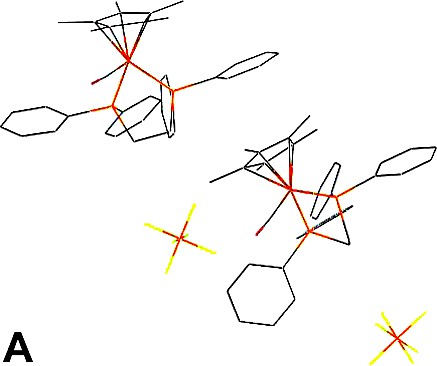
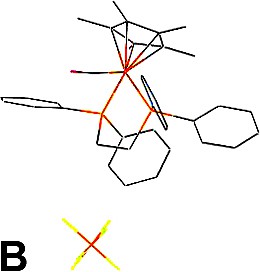
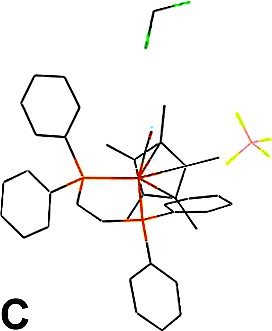
Packing for 2+PF6– (polymorph A) (a), 2+PF6– (polymorph B) (b) and 2+BF4–·CH2Cl2 (c).
Concerning the hexafluorophosphate salts, two molecules of 2+PF6– constitute the asymmetric unit arranged in a monoclinic space group in polymorph A, while only one molecule of 2+PF6– constitutes the asymmetric unit arranged in an orthorhombic symmetry in polymorph B (Tables 1 and 2). The packing of polymorph B is similar to that observed for the analogue of 2+PF6– featuring an unsubstituted cyclopentadienyl group (3+BF4–) [16]. The cationic iron(II) complexes (2+) can be considered as isolated units in both polymorphs owing to the rather large molecular separations between metal centres (> 5 Å). In the polymorph A, the molecular units manage to achieve a slightly denser packing than in polymorph B (1.456 vs. 1.384 g cm–3), as also manifested by more numerous close contacts between molecules in comparison to B. A closer investigation of the packing reveals that hydrogen bond-like interactions take place in polymorph A between the CO ligand of one molecule of the asymmetric unit and two methyl-hydrogen atoms of the cyclopentadienyl ligand of a neighbouring equivalent molecule (d(O···H) = 2.693 Å and 2.655 Å; d(O···C) = 3.580 Å and 3.545 Å) [17]. The second molecule of the asymmetric unit of A does not have similar interactions with the carbonyl ligand. However, in both polymorphs the most important contacts remain intramolecular in nature (see Section 3.1) and the hydrogen bond detected for A is possibly not determining in terms of energetic stabilisation compared to the other intra-, but also intermolecular interactions [18]. Now, when focusing more specifically on intermolecular contacts, further inspection shows that the shorter ones take place between hydrogen atoms of phenyl groups of the diphosphine ligand and fluorine atoms of the hexafluorophosphate ions.
Crystal data, data collection, and refinement parameters for 2+PF6– (A), 2+PF6– (B) and 2+BF4–
| Compound | 2+PF6– (A) | 2+PF6– (B) | 2+BF4– |
| Structural formula of the asymmetric unit | 2(C37H39O1P2Fe1, PF6) | C37H39O1P2Fe1, PF6 | C37H39O1P2Fe1, BF4, CH2Cl2 |
| Fw (g) | 1524.88 | 762.47 | 789.20 |
| Temperature (K) | 293(2) | 293(2) | 293(2) |
| Crystal system | monoclinic | orthorhombic | monoclinic |
| Space group | P21/c | P212121 | Cc |
| a (Å) | 14.1473(2) | 12.683(5) | 23.267(5) |
| b (Å) | 22.0582(3) | 16.768(5) | 8.701(5) |
| c (Å) | 22.2959(3) | 17.198(5) | 20.596 |
| α (°) | 90.0 | 90.0 | 90.0 |
| β (°) | 91.70(1) | 90.0 | 116.60(1) |
| γ (°) | 90.0 | 90.0 | 90.0 |
| V (Å–3) | 6955(2) | 3657(2) | 3728(1) |
| Z | 4 | 4 | 4 |
| Dcalc (g cm–3) | 1.456 | 1.384 | 1.410 |
| Crystal size (mm) | 0.42 × 0.38 × 0.15 | 0.60 × 0.25 × 0.07 | 1.00 × 0.90 × 0.40 |
| F(000) | 3152 | 1576 | 1532 |
| Diffractometer (NONIUS) | KappaCCD | KappaCCD | KappaCCD |
| Radiation | Mo Kα | Mo Kα | Mo Kα |
| Absorption coefficient (mm–1) | 0.634 | 0.603 | 0.684 |
| Data collection: frames, Ω rotation (°) and seconds/frame | 146, 1.5, 30 | 184, 2.0, 120 | 153, 1.8, 27 |
| Θ range | 1.30–27.51 | 3.11–26.22 | 2.20–27.48 |
| Range h, k, l | 0/18, 0/28, –28/28 | 15/15, –20/20, –21/21 | –29/30, –10/11, –26/26 |
| Number of total reflections | 58,683 | 42,466 | 18,450 |
| Number of unique reflections | 15,757 | 7248 | 7998 |
| Number of observed reflections [I > 2σ(I)] | 11,198 | 5164 | 5906 |
| Restraints/parameters | 0/866 | 0/429 | 0/461 |
| w = 1/[σ2(Fo)2+(aP)2+bP] (where P = [Fo2+Fc2]/3) | a = 0.0915, b = 6.0047 | a = 0.0441, b = 3.6641 | a = 0.086, b = 3.182 |
| Final R | 0.0545 | 0.0570 | 0.056 |
| Rw | 0.1425 | 0.1144 | 0.136 |
| R indices (all data) | 0.0870 | 0.0991 | 0.091 |
| Rw (all data) | 0.1649 | 0.1370 | 0.155 |
| Goodness of fit/F2 (Sw) | 1.002 | 1.035 | 1.027 |
| Largest difference peak and hole (e Å–3) | 0.624, –0.457 | 0.270, –0.288 | 0.588, –0.439 |
Selected bond lengths (Å) and angles (°) for 2+PF6–(A), 2+PF6–(B) and 2+BF4–
| Compound | 2+PF6–(A) | 2+PF6–(B) | 2+BF4–(A) |
| Selected bond lengths | |||
| Fe–(Cp*)centroid | 1.756a/1.750b | 1.756 | 1.766 |
| Fe–P1 | 2.2232(9)/2.2327(9) | 2.2280(16) | 2.2327(12) |
| Fe–P2 | 2.2446(9)/2.2439(9) | 2.2213(16) | 2.2424(15) |
| Fe–C37 | 1.762(3)/1.781(4) | 1.752(6) | 1.805(7) |
| C37–O1 | 1.119(4)/1.088(4) | 1.153(7) | 1.042(8) |
| Selected bond angles | |||
| P1–Fe–P2 | 85.82(3)/86.15(3) | 84.96(6) | 86.51(5) |
| P1–Fe–C37 | 90.49(10)/88.15(10) | 91.7(2) | 88.40(18) |
| P2–Fe–C37 | 87.78(10)/89.96(10) | 87.01(19) | 90.09(16) |
| Fe–C37–O1 | 175.4(3)/176.0(3) | 175.4(2) | 175.8(5) |
a Molecule 1 in the asymmetric unit.
b Molecule 2 in the asymmetric unit.
Concerning the tetrafluoroborate salt 2+BF4–, one molecule constitutes the asymmetric unit arranged in a monoclinic space group. Relatively fewer intermolecular close contacts take place between molecular units and again, the shortest contacts are mostly intramolecular. In contrast to the tetrafluoroborate ions, for which one long hydrogen bond-like interaction takes place between a fluorine atom and an hydrogen atom of the dppe ligand of 2+, the (disordered) dichloromethane solvent exhibits no close contact with any neighbouring molecular unit.
The bond distances and angles (Table 2) around the metal centre for 2+ are in the range reported for related cationic carbonyl complexes like 3+ or 4+ (Fig. 4)), the latter complex bearing the bis(diphenylphosphino)methane (dppm) ligand instead of dppe [16,19]. The Fe–P bonds are slightly longer than those commonly encountered (ca. 2.18 Å) in neutral piano-stool acetylides [14] or halides complexes [20] featuring similar coordination spheres (i.e. (η2-dppe)(η5-C5Me5)Fe(II) complexes) and are closer to these found for oxidised Fe(III) analogues (ca. 2.24 Å) [14]. These can traced back to less metal-to-ligand back-donation taking place toward the phosphine atoms in 2+ when the metal centre bears a positive charge. While the Fe–C(O) bond lengths are close to the mean value usually observed for such a bond (i.e. 1.784 Å), the C–O bond lengths are in general slightly below the mean value (1.143 Å) [21]. The comparison of the structural features of the carbonyl ligand in 2+ with these of 3+ and 4+ (Table 3), suggests that retrodonation is slightly less pronounced in the former. Indeed, except for 2+PF6–(B), the Fe–C bonds appear longer and the C–O bond distances shorter for 2+ than for 3+ and 4+ (see also Section 3).
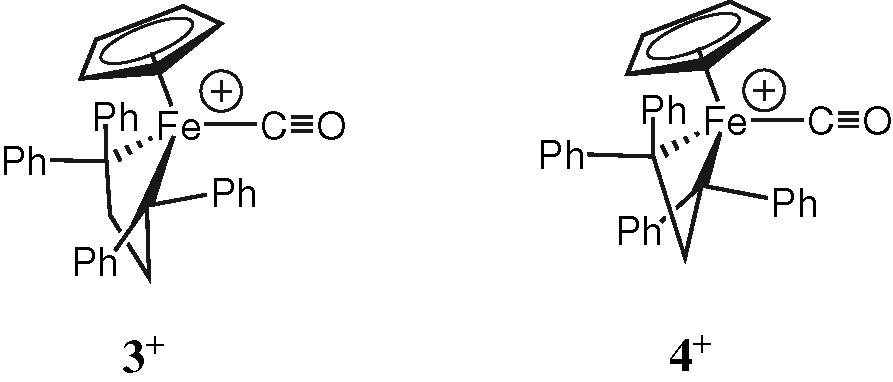
Structures of 3+ and 4+.
Structural and spectroscopic features of the carbonyl ligand in 2+ and related iron(II) piano-stool mono-carbonyl complexes
| Compound | Space group | Fe–C (Å) | C–O (Å) | Fe–C–O (°) | νCO (cm–1) | Reference |
| 2+PF6–(A) | P21/c | 1.762(3)a | 1.119(4) | 175.4(3) | 1910 (sh)c | This work |
| 1.781(4)b | 1.088(4) | 176.0(3) | 1949 (vs)c | |||
| 2+PF6–(B) | P212121 | 1.752(6) | 1.153(7) | 175.4(2) | 1934 (vs)c | This work |
| 2+BF4– | Cc | 1.805(7) | 1.042(8) | 175.8(5) | 1945 (vs)c | This work |
| 3+BF4– | P212121 | 1.744(5) | 1.139(7) | 178.6(5) | 1980 | [16] |
| 4+PF6– | P21/n | 1.744(5) | 1.145(5) | 176.7(4) | 1975 (w) | [19] |
a Molecule 1 in the asymmetric unit.
b Molecule 2 in the asymmetric unit.
c Recorded for the crystalline samples in KBr plates.
2.2 The carbonyl stretch [ν(CO)] in 2+
In order to ascertain that the isolated crystals were indeed the known carbonyl adduct 2+ and were actually different from an hypothetical isostructural [(dppe)(C5Me5)FeCN][PF6] iron(III) cyanide, their infrared spectra were recorded and compared to these of 2+BF4–. The identical carbonyl absorption at 1950 cm–1 in dichloromethane solution (Fig. 5a) establishes their identity as the cationic carbonyl adduct 2+. Surprisingly, in spite of the distance between molecular units, the infrared spectrum of the two polymorphic samples of 2+PF6– recorded in solid-state exhibit significant differences. Thus, the large red crystals (structure A) in KBr pellets or as a suspension in nujol/KBr have an absorption at 1949 ± 2 cm–1 with a very slight shoulder at ca. 1910 cm–1, while structure B has an absorption centred at 1934 cm–1 (Fig. 5b). Apparently, the packing exerts a sizeable effect on this absorption in the solid state. Comparison of ν(CO) with data available from the literature suggests that the form B was previously isolated for 2+PF6– [15]. For the tetrafluoroborate salt 2+BF4–, an absorption at 1945 ± 2 cm–1 is observed in KBr pellets made from the crystals. Obviously, a change in the counter ion has not a large influence on ν(CO). The value (1947 ± 2 cm–1) obtained for a powdered (re-precipitated) sample of 2+BF4– suggests that the solid state structure is preserved in ‘amorphous’ samples. All the distinct absorptions obtained for various solid samples of 2+ merge into a single absorption upon dissolution in dichloromethane, indicating also that no specific interaction takes place between 2+ and its counter ion in this solvent. Without surprise, the ν(CO) recorded in solution for 2+, is lower in energy than for 3+ or 4+. This is in line with the more electron-releasing nature of the permethylated cyclopentadienyl ring in 2+ and indicates that more retrodonation takes place in this compound than in 3+ or 4+, which feature a simple cyclopentadienyl ligand. However, as mentioned above, the structural data seem to indicate the opposite trend. We will return to this question later on (see the Section 3). Finally, these solid samples do not show any clear ν(CO) bands in their Raman spectra, indicating that the carbonyl stretch does not induce a marked polarisability change in these compounds.
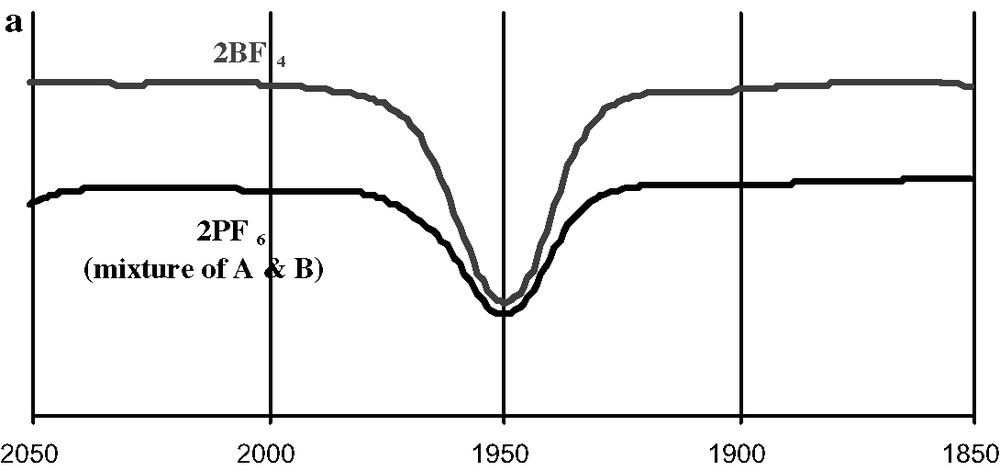
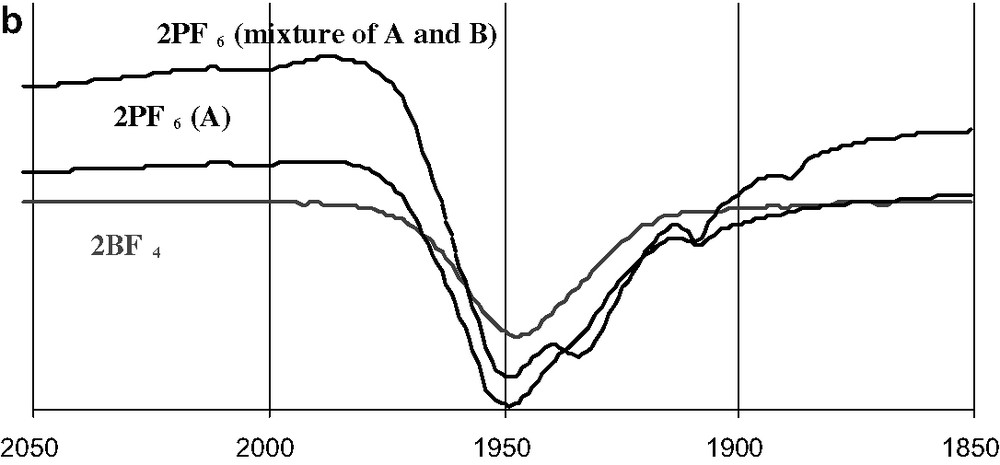
Infrared spectra (cm–1) of 2+ samples in CH2Cl2 solution (a) and in the solid state (b).
The infrared spectrum of the crystalline material isolated for 2+PF6– also reveals the conspicuous absence of the starting 1+PF6–, which presents a characteristic ν(C≡C) around 1962 cm–1 and the characteristic A1 phenyl stretch at around 1588 cm–1. While the former absorption could have been partly hidden by the carbonyl absorption, the later is clearly distinct from these of compound 2+ and was also absent. Thus, most of 1+PF6– has apparently been converted in 2+PF6– in ca. 4 weeks.
2.3 Reaction of 1+ with molecular oxygen
Among the various hypotheses that can be envisioned for rationalising the formation of 2+PF6– from 1+PF6–, we checked the most likely possibilities: reactions with traces of molecular oxygen or with moisture. Thus, a sample of 1+PF6– was dissolved in dichloromethane and exposed to dry oxygen. The reaction was then monitored by 1H and 31P NMR. No fast reaction is observed, however, based on the intensity of the NMR signals, more than 90% of the starting 1+PF6– is actually converted into 2+PF6– after 36 h. The reaction is very clean, since essentially 2+PF6– and a single organic product of mass 397 ± 1 are present. Its assignment as bis(4-N,N′-dimethylamino)dibenzyl (5) is supported by the NMR [22] and infrared data. NMR and GC-MS of an original sample of 4-(dimethylamino)benzophenone also indicated that this product was not present.
The disappearance of 1+PF6– could conveniently be monitored by UV spectroscopy using a specially adapted cell (tonometer) presenting a gas-reservoir in presence of different partial pressures of oxygen (ppO2)1. This is a first order process in 1+PF6– as demonstrated by the linear plots obtained when the logarithm of absorbance at 880 nm is plotted against time (Fig. 7a). A half life of ca. 5 h 30 mn for 1+PF6– under atmospheric conditions (ppO2 = 0.2 bar) is found. A linear plot is also obtained when the experimental apparent decay constants (kapp) obtained for the different partial pressures are plotted versus these partial oxygen pressures (Fig. 7b). This indicates that the reaction rate is also first order in oxygen concentration, obeying a second order rate equation like (1).
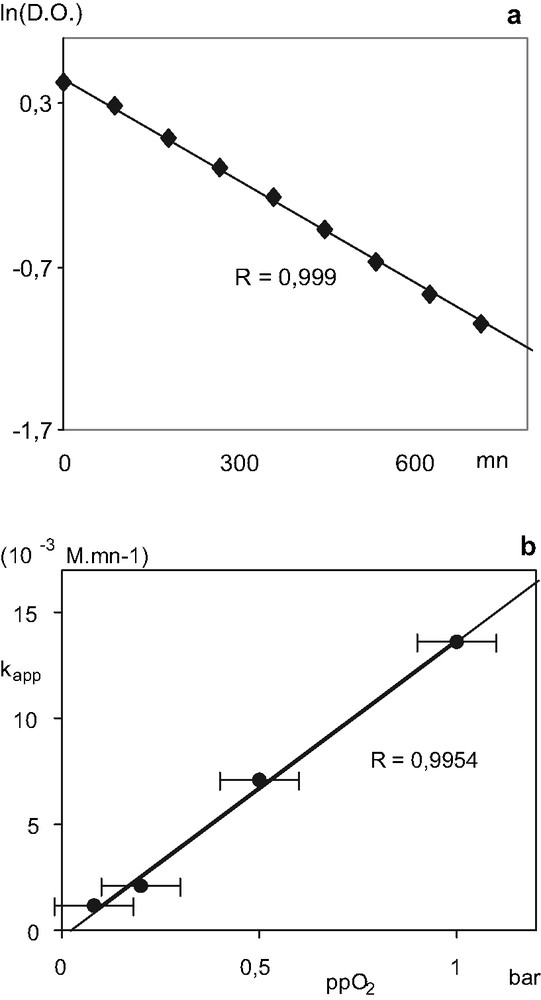
Kinetic data. (a) Plot of the evolution of the logarithm of the optical density at 880 nm for 1+PF6– vs. time for ppO2 = 0.2 bar. (b) Plot of the various kapp determined vs. corresponding ppO2. Read R2 instead of R, D.O. instead of O.D., and 103 M min–1 instead of 10–3 M.mn–1.
–d[1+]/dt = k[1+][O2] (1)
Control experiments revealed that over the same period of time, 1+PF6– is comparatively stable under argon in neat dichloromethane (less than 10% decomposition). The decomposition is also not significantly accelerated when water was introduced in the medium. Thus, the reaction of 1+PF6– with water is much slower than that with dioxygen.
Finally, we have stated that 1+PF6– reacts faster with oxygen than an analogous Fe(III) complex like 6+PF6– bearing the strongly electron-withdrawing NO2 group in place of NMe2 (Fig. 6). With the later complex, under atmospheric conditions (ppO2 = 0.2 bar), the reaction of 6+PF6– is roughly 15 times slower than for 1+PF6– (kapp = 4 × 10–5 M min–1 vs. kapp = 2.1 × 10–3 M min–1).
Structures of 6+ and 7+.
3 Discussion
3.1 Structural vs. spectroscopic features of the carbonyl ligand
Very good correspondence has previously been evidenced by Riley and Davies [16] between structural data and carbonyl absorptions for piano-stool cyclopentadienyl carbonyl Fe(II) complexes like 3+. In accordance with the Dewar–Chatt–Duncanson description of synergic bonding between iron and CO, the available structural data suggested that stronger force constants for the carbonyl ligand would actually correspond to shorter CO bonds in the solid state. This trend was also obeyed by the spectroscopic and structural features reported later on by Ruiz et al. [19] for the closely related cationic iron(II) complex 4+ featuring the bis(diphenylphosphino)methane (dppm) ligand in place of dppe (4+PF6–) (Table 3). However, this picture does not agree anymore with the data gathered here for the various samples of 2+. Indeed, the data in Table 3 clearly show that the solid state carbonyl absorptions do significantly depend on the crystalline arrangement and therefore do not provide a truly ‘molecular’ information for these compounds. In addition, several inconsistencies between CO-bond lengths and the corresponding solid state carbonyl absorptions have been noted, when analysed in this context2. Thus, the data strongly suggest that the correspondence between CO-bond lengths and stretching frequencies previously observed by Riley and coworkers is not a general feature for piano-stool iron(II) carbonyl complexes like 2+. Looking for explanations of this fact, we could put forward two statements.
(i) First, the CO-bond length/ν(CO) correspondence is based on the assumption that the ν(CO) does actually correspond to a pure (uncoupled) stretching fundamental (or at least that its vibronic coupling with adjacent vibrators is not modified along the series). In the samples of 2+ presently characterised, the separation between the carbonyl ligands precludes any intermolecular vibronic coupling, nevertheless, changes due to (through-space) intramolecular vibronic coupling could intervene.
(ii) The second reason evidently relates to the precision of the bond lengths determined in our measurements. Braga and Koetzle [24,25] have stated some years ago that unless very high quality data was obtained, structural data obtained with X-ray on carbonyl ligands is often plagued by systematic errors in true atomic positions, which are not estimated in the computed esds. This is because conventional treatment of X-ray data does not take in consideration the possible deformation of the electronic clouds occasioned by molecular vibrations or librations. An indication of the magnitude of such an effect is given by the <ΔC–O> and <ΔM–C> indices reflecting the relative difference in atomic coordinates of the carbonyl ligand atoms when passing from isotropic to anisotropic refinement. In this connection, <ΔC–O> and <ΔM–C> indices determined for the various cations 2+ (Table 4) suggest that the experimental CO-bond distances are significantly affected by this phenomenon in comparison to other carbonyl complexes [24].
Intramolecular close-contacts below van der Waals radii between the C(O) atom and selected atoms (Å) and <ΔC–O> (Å2) in 2+
| Molecular unit | Cortho1a | Cortho2b | Hc | <ΔM–C> | <ΔC–O> |
| 2+PF6–(A)d | C18(3.046) | C32(3.295) | / | 0.0153 | –0.0065 |
| 2+PF6–(A)e | C70(3.146) | C64(3.280) | H64(2.888) | 0.0076 | –0.0086 |
| 2+PF6–(B) | C26(3.196) | / | H12A(2.679) | 0.0080 | –0.0055 |
| H50B(2.662) | |||||
| 2+BF4– | C32(3.220) | / | H32(2.795) | 0.0367 | –0.0142 |
a Numbering (distance in Å) of the nearest ortho carbon atom of the nearby dppe phenyl group.
b Numbering (distance in Å) of the nearest ortho carbon atom of the other phenyl group.
c Numbering (distance in Å) to hydrogen atoms on phenyl groups of the dppe ligand or on methyl groups of the C5Me5 ligand.
d Molecule 1 in the asymmetric unit.
e Molecule 2 in the asymmetric unit.
Thus, we suggest that some deviation of the CO stretch from a linear motion along the metal–carbon–oxygen axis might presently be induced by the large steric strain resulting from the simultaneous presence of the dppe and C5Me5 ligands in the coordination sphere of the iron centre. Indeed, atomic separations significantly less than van der Waals contacts take place between the carbon atom of the carbonyl ligand (C37) and ortho-carbon atoms belonging to nearby phenyl groups of the dppe ligand, especially in polymorph A (Table 4). These might also be connected to the pronounced bending of the carbonyl ligand observed for the various samples of 2+ (Fe–C–O angle in Table 3) in comparison to 3+ and 4+. Whether this phenomenon results from a simple steric repulsion or involves some kind of through-space vibronic coupling between the CO stretch and the C–C and C–H motions of the nearby vibrators are presently unknown. Nevertheless, it would nicely explain the breakdown in the CO-bond length/ν(CO) correspondence stated for 2+ by rendering the carbonyl stretch highly conformation dependant in the solid state.
3.2 Proposed mechanism for the oxidation of 1+
Our kinetic investigations indicate that the formation of samples of 2+PF6– during attempts to crystallise 1+PF6– most probably results from reaction of the latter with traces of oxygen dissolved in the solvents. The lack of reactivity with water suggests that the process has only limited similarity with the known hydrolysis of alkynes [26]. Indeed, the kinetic data strongly suggest that the present transformation is actually initiated by molecular oxygen and not by nucleophilic attack of water on any intermediate [27]. The slow step of this transformation apparently follows a second order kinetic law (Eq. 1). In accordance with these observations, we propose a radical mechanistic pathway to explain the formation of 2+PF6– (Fig. 8). Such a radical mechanism is in line with the radical cation nature of 1+PF6–. The relatively high stability of benzylic type-radicals like 8 possibly acts here as a driving force for the reaction, the slow step being the reaction between 1+ and O2. In the proposed mechanism, transient intermediates (i1–i2) have been advanced. Whether these have an actual existence or are just different stages of a transition state resulting from a direct transformation of 1+ into 2+ cannot be determined at this point. Notably, the present mechanism might also have been initiated by reaction of the dioxygen with the iron(III) centre, forming a reactive 19-electron Fe(IV) superoxo species, which would subsequently undergo ring closure to form a transient five membered metallacyclic intermediate which rearranges into i1. The formation of reactive 19-electron intermediates from 17-electron radicals has precedence in the literature [28].
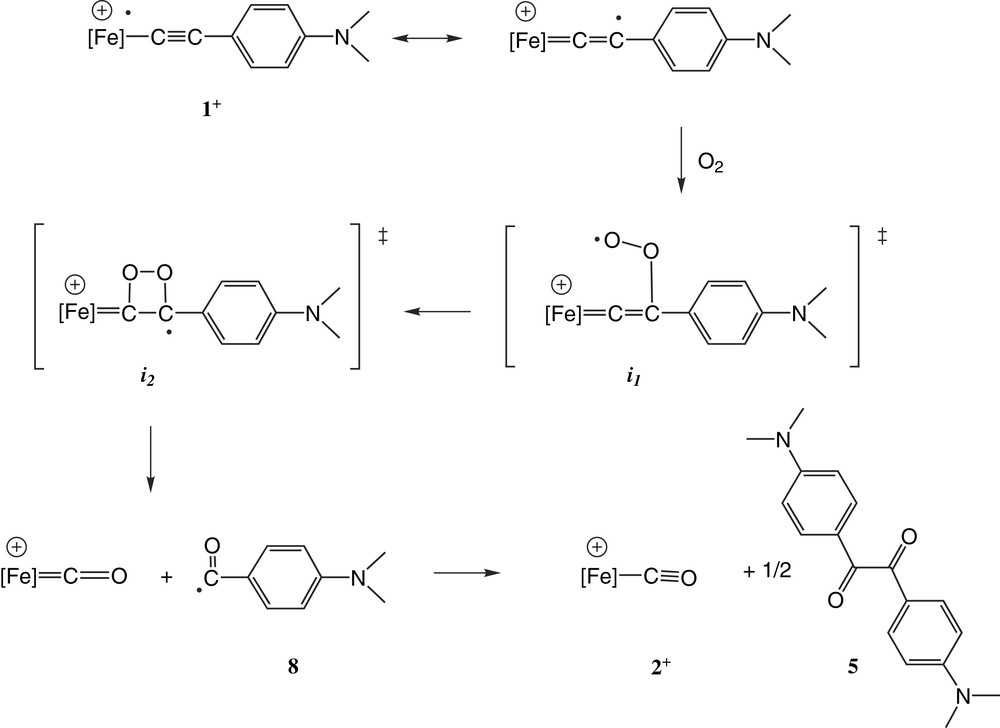
Proposed mechanism for the transformation of 1+ into 2+.
To the best of our knowledge, similar reactions have never been reported between metal acetylides and oxygen, although related oxygen transfer processes between carbon monoxide and acetylides catalysed by transition metal complexes are known [29,30]. A very close reaction between vinylidenes and molecular oxygen has been reported by Bruce in 1975, yielding benzoic acid [31]. However, in Bruce's mechanistic proposal, benzaldehyde was the primary organic product of the cleavage reaction, which was oxidised to benzoic acid in a subsequent step. In the present case, exposure to air or water does not speed up the process and no 4-(dimethylamino)benzaldehyde was detected. The intermediacy of the vinylidene 7+PF6– (Fig. 6) in the process seems, therefore, unlikely. In addition, the kinetic data clearly indicate that irreversible conversion to the vinylidene can not be the slow step of the transformation since such a process does not obey to the rate law (Eq. 1). NMR monitoring reveals also that vinylidene is not appreciably formed from 1+ over time scales allowing complete consumption of this complex in presence of oxygen, even when excess water had been introduced in the medium, excluding thereby 7+PF6– as a long lived intermediate in the reaction.
Considering that intractable mixtures often result when solutions of metal acetylides possessing unpaired electrons are exposed to air, the reaction of 1+PF6– with molecular oxygen is remarkably clean. In other words, this ‘oxidation’ process competes very efficiently with more traditional radical coupling reactions undergone by organoalkynyl radical cations [32], like hydrogen capture leading to the corresponding vinylidene or dimerisation leading to the bis-vinylidene complexes [33]. The selectivity of this process might presently originate from strong unpaired spin density on both the α- and β-carbon atoms of the alkynyl linker, which is favoured by the electron-releasing character of the dimethylamino substituent [14,34]. Notably, such a reaction constitutes an interesting example of an oxidation process of a Fe(III) complex (1+) resulting eventually in its quantitative conversion into a reduced Fe(II) species (2+). It is possibly at the origin of the pervasive observation of 2+ during FAB- or LSI-MS characterisation of electron-rich metal acetylide complexes when the compounds are introduced in non-degassed matrixes.
4 Conclusion
Two different crystalline arrangements of the known carbonyl complex 2+PF6– were isolated from the acetylide Fe(III) radical cation 1+PF6– and characterised by X-rays. The known tetrafluoroborate analogue (2+BF4–) has also been independently synthesised and structurally characterised. Both polyphorphs of 2+PF6–(A and B) and 2+BF4– exhibit distinct carbonyl absorptions and also distinct structural features for the carbonyl ligand in the solid state. However, several inconsistencies are found between these structural variations and the corresponding ν(CO). We propose that the reason is basically intramolecular in origin and results from steric bulk around the metal centre, thereby indirectly reflecting the changes in packing of the iron cations in the cell. Structural features should therefore not be taken at face value for discussing the bonding of the carbonyl ligand in complexes like 2+ at the molecular level. More importantly, we establish here that this complex forms after reaction from 1+PF6– with oxygen. This confirms that in spite of their decreased electron-richness relative to their Fe(II) redox parents, Fe(III) functional phenylalkynyl radical cations are still reactive species toward molecular oxygen, especially when substituted with electron-releasing groups in the 4-position of the aryl ring. Formation of species like 2+PF6– should therefore not be overlooked when working with related radical transition metal acetylides, especially considering the possible confusion between the carbonyl stretch in 2+ and the alkynyl stretch of Fe(III) acetylide complexes [12].
5 Experimental
5.1 General
Unless otherwise specified, all reagents and solvents were purchased from commercial suppliers and used without further purification. Distilled and degassed solvents were used for the various crystallizations. The complexes [(η2-dppe)(η5-C5Me5)FeC≡C-1,4-(C6H4)NMe2][PF6] (1+PF6–), [23] [(η2-dppe)(η5-C5Me5)FeC≡C-1,4-(C6H4)-NO2][PF6] (6+PF6–) [11] and [(η2-dppe)(η5-C5Me5)FeCO][BF4] (2+BF4–) [35] were obtained following reported syntheses. 1H NMR spectra were obtained on a BRUCKER ADVANCE 200 (200 MHz) spectrometer. 1H and 31P NMR chemical shifts are reported in units of parts per million (ppm) relative to residual protiated solvent and H3PO4 respectively. Transmittance-FTIR spectra were recorded using a Bruker IFS28 spectrometer (400–4000 cm–1). Raman spectra of the solid samples were obtained by diffuse scattering on the same apparatus and recorded in the 50–4000 cm–1 range (Stokes emission) with a laser excitation source at 1064 nm (25 mW) and a quartz separator with a FRA 106 detector. UV–Vis spectra were recorded on an UVIKON XL spectrometer. Cyclic voltammograms were recorded using a PAR 263 apparatus in CH2Cl2 (0.1 M (n-Bu)4N+ PF6–) at 25 °C, at a platinum electrode, using a SCE reference electrode and ferrocene as internal calibrant (0.460 V) [36].
5.1.1 [(η2-dppe)(η5-C5Me5)Fe–C≡O][BF4] (2+BF4–)
IR (ν, KBr/CH2Cl2, cm–1) 1947/1950 (s, C≡O). NMR 31P{1H} (δ, CDCl3, 81 MHz, ppm) 88.4 (s). NMR 19F{1H} (δ, CDCl3, 188 MHz, ppm) –151.8 (s, BF4–). NMR 1H (δ, CDCl3, 200 MHz, ppm) 7.54 (m, 20H, HAr/dppe); 2.38 (m, 4H, CH2dppe); 1.48 (s, 15H, C5(CH3)5). NMR 13C{1H} (δ, CDCl3, 50 MHz, ppm) 218.2 (t, 2JCP = 25 Hz, Fe–C≡O); 135.0–128.5 (m, CAr/dppe); 95.6 (s, C5(CH3)5); 30.5 (m, CH2/dppe); 9.8 (s, 1JCH = 128 Hz, C5(CH3)5). E0 (ΔEp, ipa/ipc) 1.39 V (0.08, 1) V vs. S.C.E. UV–Vis. (CH2Cl2) λmax (ε/103 M–1 cm–1) 320 (sh, 4.2); 380 (sh, 1.4). Mössbauer (mm s–1 vs. Fe, 2PF6, 80 K): IS 0.196, QS 1.842 [37].
5.2 Crystallography
Crystals of 2+PF6–(A) and 2+PF6–(B) were obtained by slow diffusion of n-pentane in a dichloromethane solution of 1+PF6–, while crystals of 2+BF4– were obtained by slow diffusion of n-pentane in a dichloromethane solution of an authentic sample of the 2+BF4– complex. The samples were measured on a NONIUS KappaCCD with graphite monochromatised Mo Kα radiation. The cell parameters are obtained with Denzo and Scalepack with 10 frames (omega rotation: 1° per frame) [38]. Data collection (number of frames, omega rotation, seconds per frame and h, k, l range as given in Table 1) and reduction with Denzo and Scalepack gave the independent reflections. The structures were solved with SIR-97, which revealed the non-hydrogen atoms [39]. The whole structures were then refined with SHELXL-97 by the full-matrix least-square techniques (use of F2 square magnitude; x, y, z for Fe, P, C, N and/or O atoms, x, y, z in riding mode for H atoms with variables ‘N(var.)’, observations and ‘w’ used as defined in Table 1) [40]. ORTEP views of 2+PF6–(A), 2+PF6–(B) and 2+BF4 were realised with PLATON98 [41]. The <ΔC–O> and <ΔM–C> indices have been computed using the THMA11 program [42]. Data for this structure have been deposited with the Cambridge Crystallographic Data Centre as Supplementary publication Nos CCDC-229660, 265780 and 255332. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-1223-336-033; e-mail: deposit@ccdc.cam.ac.uk).
5.3 Reaction of 1+PF6– with molecular oxygen
5.3.1 31P and 1H NMR monitoring
A 9 mg sample (0.010 mmol) of the paramagnetic complex 1+PF6– was dissolved in CD2Cl2 (1 ml) and exposed to an oxygen atmosphere. The 31P and 1H NMR spectra of the reaction medium after 48 h revealed the appearance of two diamagnetic products, identified as the cationic carbonyl complex 2+PF6– and bis(4-N,N′-dimethylamino)dibenzyl (5) in a 2:1 ratio. The presence of these products was confirmed by infrared analysis of the sample after evaporation of the solvent, while the identity of 5 was definitively established by EI-MS analysis of an aliquot of the crude sample, after filtration through silica to remove any inorganic species (see supplementary material). Under similar conditions, the same reaction was also carried out with 6+PF6–.
The reaction between 1+PF6– and H2O (1 μl; 0.056 mmol) instead of oxygen was also monitored and did not produce marked changes in the reaction medium. Typically, after the time where reaction of 1+PF6– was complete under an oxygen atmosphere, less than 30% have reacted in presence of water. Traces of an unknown product (singlet at 91.4 ppm), of the vinylidene complex (7+PF6–) corresponding to 1+PF6– (singlet at 89.5 ppm) and of the carbonyl complex 2+PF6– (singlet at 88.6 ppm) are observed in the reaction medium by 31P NMR after this time, but 1+PF6– remains clearly the dominant species.
5.3.2 UV monitoring
In a specially designed UV-cell (tonometer) with an adapted gas-reservoir of 125 ml, a solution of 1+PF6– (2 mg, ca. 9.1 × 10–5 M) in CH2Cl2 was exposed to oxygen–argon or oxygen–nitrogen mixtures containing increasing proportions of oxygen (10–25–62–125 ml) and the decay of the characteristic absorbance of 1+PF6– and 6+PF6– was monitored over time.
6 Supporting information available
Full details of the X-ray structure of 2+PF6– (Polymorphs A and B) and 2+BF4– including tables of atomic positional parameters, bond distances and angles, anisotropic and isotropic thermal displacement parameters. GC-MS trace and NMR spectra 1+PF6– after reaction of with dioxygen (ppO2 = 0.2 bar) (32 pages). Ordering information is given on any current masthead page.
Acknowledgements
We thank C. Ruzié (UMR 6509) for kindly providing the UV-cell (tonometer) used in the kinetic measurements and Dr. J.-R. Hamon (UMR 6509) for helpful discussions. The French CNRS is acknowledged for financial support.
1 In line with the data reported for [(η2-dppm) (η5-C5H5)FeCO][PF6], [19] the carbonyl adduct 2+ exhibits only a weak absorption at 380 nm in its UV spectrum with a tail extending in the visible range. Accordingly, the red cubes (structure A) or pale-orange plates (structure B) of 2+PF6– give pale yellow solutions upon dissolution in dichloromethane that become more and more red upon concentration. In contrast, 1+PF6– is much more strongly coloured in reason of a strong absorption at 880 nm in dichloromethane [23].
2 Also, the CO-bond length/ν(CO) correspondence does not hold among the various molecules of 2+ constituting the asymetric units in these polymorph (Table 3). For instance, the molecules in 2+PF6–(A) present a significantly shorter bond length than that of 2+PF6–(B) (1.119 ± 0.012 Å and 1.088 ± 0.012 Å vs. 1.153 ± 0.021 Å) and should therefore exhibit both higher wave numbers for the carbonyl stretch. Notably, this deviation cannot be ascribed to the existence of hydrogen-bond interactions involving the carbonyl ligand of one of the molecules of the asymmetric unit in 2+PF6–(A), since the ligand in this molecule curiously exhibits the shorter C–O bond (1.088 Å) [18].