

1 Introduction
Since the spectroscopic studies of the d8−d8 metal complex [Pt2(POP)4]4− [1,2], there has been immense interest in the investigation of weak metal···metal interactions and metal−metal bond establishment in the excited state in transition metal complexes. The fascinating photophysical and photochemical properties [3–8] as well as the diverse coordination modes [8] exhibited by the related d10 transition metal complexes have also made them ideal candidates for the exploration and understanding of the photochemical properties and emission origins of this kind. In accord, a number of luminescent d10 species have been reported to derive their emission from pure metal-centred d−s or d−p excited states originating from the weak metal···metal interactions [3,4,9–11]. On the other hand, there are examples of polynuclear d10 metal complexes, which have been suggested to emit from metal cluster-centred d−s/XLCT or d−s/LMCT mixed excited states other than a pure metal-centred origin [8,9].
Other than the d10 system, d8 and d6 transition metal alkynyl complexes have also aroused enormous attention in recent years owing to their intriguing properties [12–14]. In particular, d8 square-planar platinum(II) systems with diimine ligands have shown rich photophysical, polymorphic and solvent-induced aggregation properties [15–19]. The electronic properties of this class of materials have been reported to be closely related to the extent of weak Pt···Pt interactions, or the microenvironment about the platinum metal centres [15–19]. Thus, it is envisioned that the molecular approach to the design and assembly of luminescent metal-based materials would be a promising tool for the exploration of the spectroscopic origins of complexes with mixed metal centres as well as those with weak metal···metal interactions.
2 Metal chalcogenides and pnictogenides
2.1 Tetranuclear copper(I) and silver(I) chalcogenides and pnictogenides
A series of tetranuclear copper(I) and silver(I) complexes with μ4-bridging chalcogenides and pnictogenides, [M4(μ-P∧P)4(μ4-E)]2+ [M = Cu, E = S, Se, PPh; M = Ag, E = S, Se, Te; P∧P = bis(diphenylphosphino)methane (dppm), bis[di(4-methylphenyl)phosphino]methane], have been prepared in our laboratory (Fig. 1) [20–25]. In general, the four metal atoms in these complexes are arranged in a distorted rectangular array and are bridged by a chalcogenido or pnictogenido ligand atop of it. The apices of the bridging diphosphine ligands are not pointing to the same direction but are in an alternating up and down saddle-like fashion.
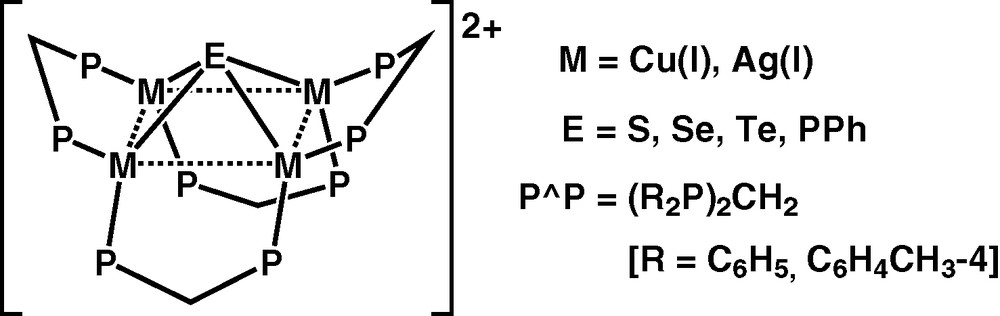
Tetranuclear copper(I) and silver(I) chaleogenides and pnictogenides.
These complexes are found to be soluble in common organic solvents and possess rich photophysical properties. The electronic absorption spectra of this class of complexes generally showed a high-energy absorption shoulder at ca. 240−260 nm with an extinction coefficient in the order of 104 dm3 mol−1 cm−1, and a lower energy absorption tail in the region of 350−450 nm. The emission energies of these complexes are found to be dependent on both the chalcogenido or pnictogenido ligand and the metal centre. Red shifts in the solid-state emission energies are observed when changing only the chalcogenido or pnictogenido ligand from S (P^P = dppm; M = Cu, 579 nm; M = Ag, 516 nm) to Se (P^P = dppm; M = Cu, 595 nm; M = Ag, 527 nm) to Te (P^P = dppm; M = Ag, 574 nm) to PPh (P^P = dppm; M = Cu, 718 nm), as well as changing only the metal centres from Ag4 (P∧P = dppm; E = S, 516 nm; E = Se, 527 nm) to Cu4 (P∧P = dppm; E = S, 579 nm; E = Se, 595 nm). Thus, the emission of these copper(I) and silver(I) clusters with bridging chalcogenido or pnictogenido ligands are assigned to originate predominantly from a ligand-to-metal charge transfer LMCT (E2− → M4) triplet state, probably mixed with a Cu(I) or Ag(I) metal-centred MC (d−s or d−p) state, modified by weak metal···metal interactions.
The phosphorescent states of these complexes are found to be quenched by a series of pyridinium acceptors via an outersphere electron transfer process, which is indicative of their highly reducing nature in the excited states. For example, an excited-state reduction potential, E°([Cu4]3+/[Cu4]2+*) of −1.55(10) V vs. S.S.C.E. has been estimated for [Cu4(μ-dppm)4(μ4-Se)]2+ with a reorganisation energy λ of 1.12(10) eV. Transient absorption difference spectrum recorded for [Cu4(μ-dppm)4(μ4-Se)]2+ in the presence of a pyridinium acceptor showed absorption bands at ca. 390, 484 and 693 nm which is characteristic of the pyridinyl radical, and inter-valence charge transfer (IVCT) bands for [CuICuICuICuII], respectively [26].
2.2 High nuclearity gold(I) chalcogenides
In addition to the tetranuclear copper(I) and silver(I) chalcogenide complexes, a series of high nuclearity gold(I) chalcogenide clusters have been isolated in our laboratory via the reactions of the dinuclear gold(I) complexes, [Au2(μ-P∧P)Cl2] (P∧P = dppm and Ph2N(nPr)PPh), with H2S in a mixture of ethanol and pyridine (Fig. 2) [27–29]. The resulting multinuclear complexes with chalcogenido ligands, [Au10{μ-Ph2PN(nPr)PPh2}4(μ3-S)4]2+, [Au6{μ-Ph2PN(p-CH3C6H4)PPh2}3(μ3-S)2]2+ and [Au12(μ-dppm)6(μ3-S)4]4+, are found to be strongly emissive both in the solid-state and in fluid solutions. The lowest-energy excited state of this class of complexes are assigned as derived from an admixture of ligand-to-metal charge transfer LMCT[E2− → Au(I)] and metal-centred MC(d → s/d → p) states, modified by weak Au···Au interactions [27–29].
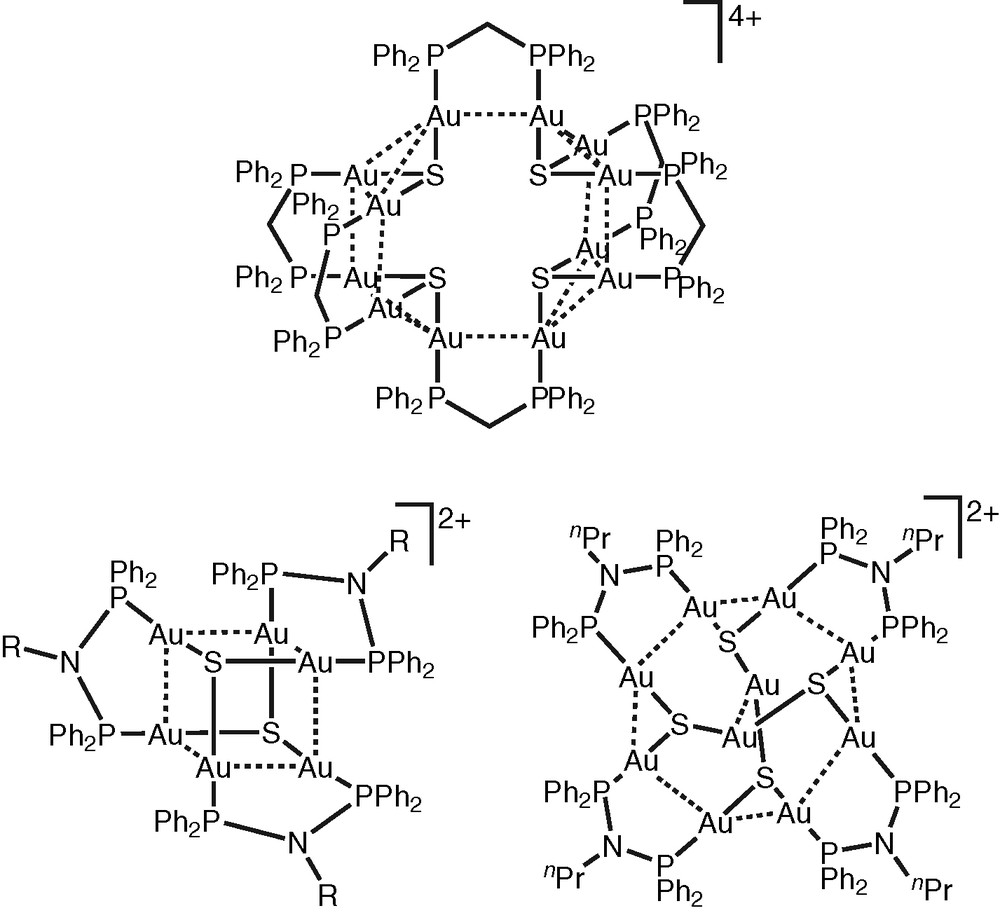
High-nuclearity gold(I) chaleogenides.
3 Building blocks for luminescent organometallic oligomers and dendrimers
3.1 Polynuclear copper(I) and silver(I) alkynyl complexes
Besides the copper(I) and silver(I) complexes with chalcogenido and pnictogenido ligands, a series of alkynyl complexes with copper(I) and silver(I) metal centres have also been successfully synthesised by us (Fig. 3) [30–35]. The electronic absorption spectra of the triangulo-M3 complexes, [M3(μ-P∧P)3(μ3-η1-C≡CR)2]+ and [M3(μ-P∧P)3(μ3-η1-C≡CR)]2+ [M = Cu(I), Ag(I); R = aryl, alkyl; P∧P = dppm; Ph2N(R′)PPh2, R′ = Ph, nPr, C6H4CH3-4, C6H4F-4], are dominated by the intraligand absorption bands of the diphosphine and alkynyl ligands but with a low-energy shoulder tailing to ca. 400 nm.
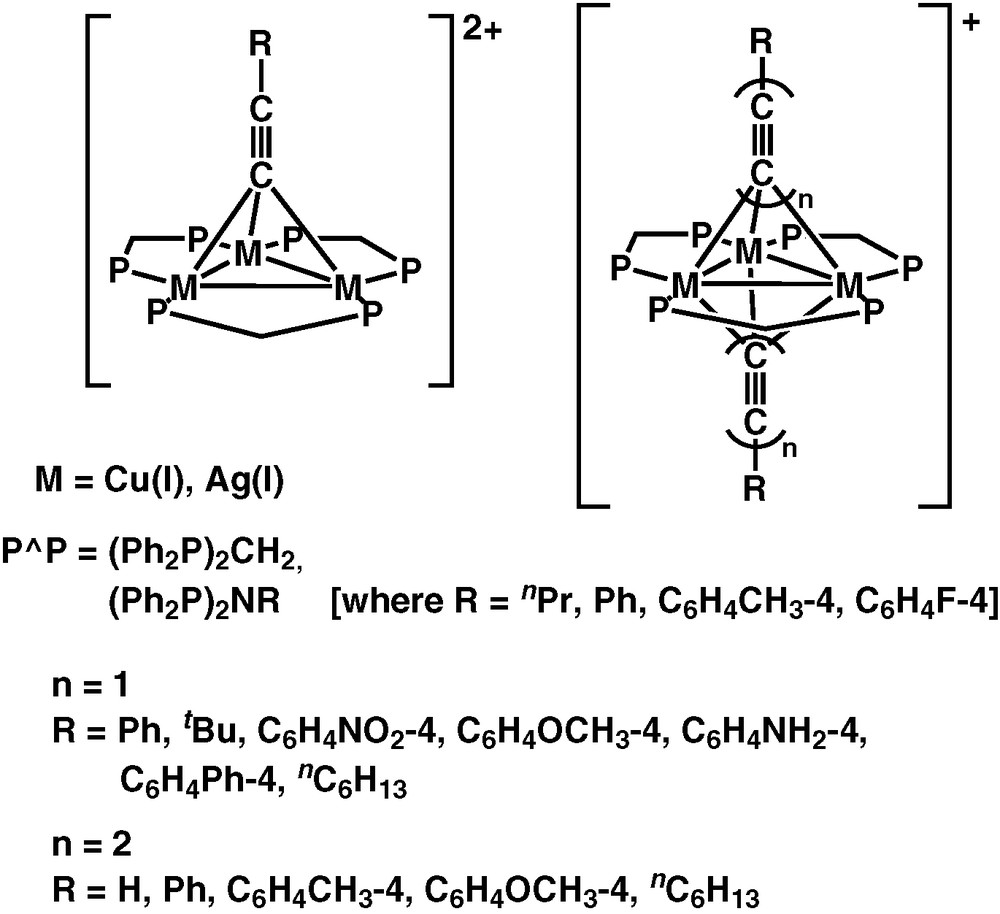
Polynuclear copper(I) and silver(I) alkynyl complexes.
Long-lived and intense luminescence is observed upon photo-excitation of the complexes in the solid-state or in fluid solutions. Similar to the copper(I) and silver(I) chalcogenido and pnictogenido complexes, the emission energies of these complexes are also found to be dependent on the nature of the ligand and the metal core. For instance, the emission energies of the monoynyl complexes [Cu3(μ-dppm)3(μ3-η1-C≡CR)]2+ in acetone solution show an order of: R = C6H4–OCH3-4 (483 nm) ≈ Ph (499 nm) > C6H4–NH2-4 (504 nm) > tBu (640 nm) ≈ nC6H13 (650 nm), which is in line with the increasing electron-donating ability of the alkynyl ligand. Red shifts of emission energies are also noted when going from Ag(I) to Cu(I) complexes with identical diphosphine and alkynyl ligands. These observations are indicative of substantial involvement of a ligand-to-metal charge transfer LMCT[C≡C → Cu3] character in their emissive state. However, in view of the presence of short M···M distances in these complexes, especially for the bicapped system, mixing of a certain degree of metal-centred MC[d–s] character is also possible. Therefore, the lowest-lying emissive state of these complexes is best described as an admixture of 3LMCT[C≡C → Cu3]/3MC[d–s] character modified by weak M···M interactions [30–35]. As an extention of this work, a series of trinuclear Cu(I) oligoynyl complexes, [Cu3(μ-dppm)3{μ3-η1-(C≡C)nR}2]+ (n = 1, 2; R = H, Ph, C6H4–CH3-4, C6H4–OCH3-4) have recently been studied by us [36]. The emission spectra of the complexes show emission bands with vibrational progressional spacing of ca. 1811−2058 cm−1, which are characteristic of ν(C≡C), and the emission energy is dependent on the nature of the substituents on the diynyl ligand (Fig. 4). Thus, the low-lying 3LMCT emissive state for these oligoynyl complexes are likely to be mixed with the 3IL[π–π*] of the oligoynyl moieties [36].
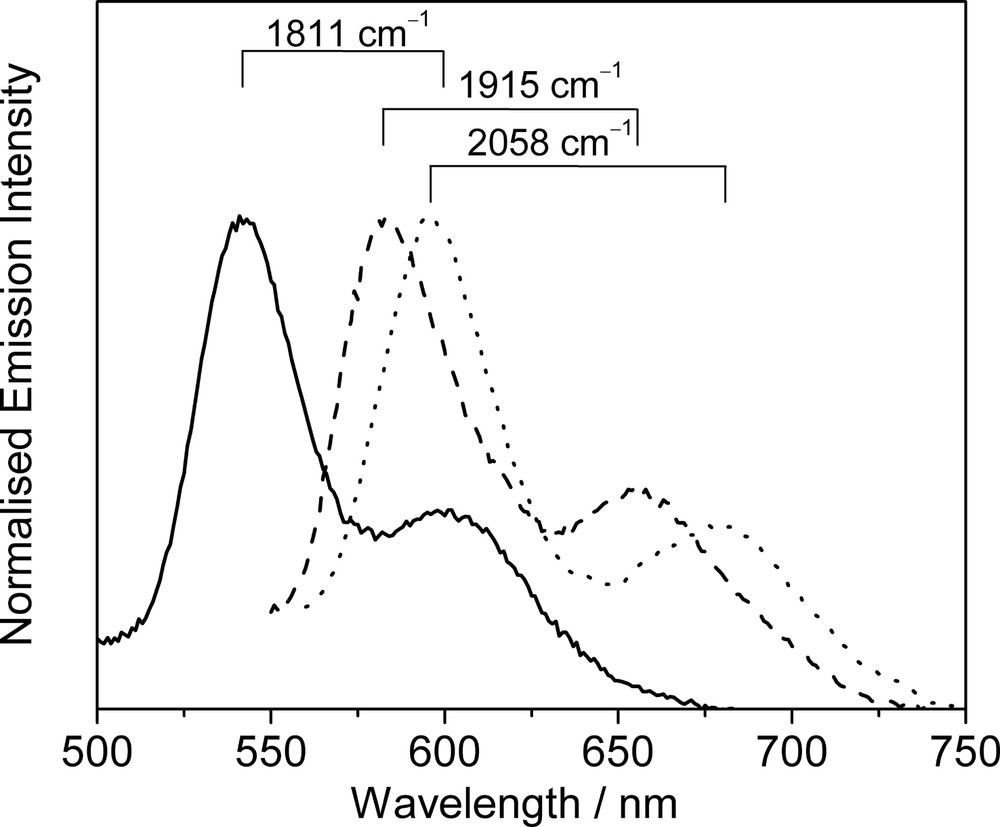
Normalised emission spectra of [Cu3(μ-dppm)3(μ3-η1-C≡C−C≡CR)2]+ [R = Ph (—), C6H4–CH3-4 (- - -) and C6H4–OCH3-4 (·····)] in the solid-state at 298 K.
In addition to the trinuclear Cu(I) and Ag(I) alkynyl complexes, a series of tetranuclear Cu(I) and hexanuclear Cu(I) and Ag(I) alkynyl complexes have been synthesised in our laboratory. The tetranuclear Cu(I) alkynyl complex, [Cu4(μ-dppm)4(μ4-η1,η2-C≡C–)]2+, was synthesised by the reaction of [Cu2(μ-dppm)2(CH3CN)2]2+ with Me3SiC≡CH in the presence of nBuLi in THF under an inert atmosphere of nitrogen [37]. Interestingly, the C≡C2− moiety in this tetranuclear complex is found to oscillate inside the distorted rectangular array of four copper atoms with a ΔG‡ value of ca. 38 kJ mol−1 [38]. On the other hand, reaction of the dinuclear complex [M2(μ-dppm)2(CH3CN)2]2+ (M = Cu, Ag) with 1,4-diethynylbenzene in the presence of nBuLi in THF afforded the respective diynyl-bridged hexanuclear Cu(I) and Ag(I) complexes, [M3(μ-dppm)3(μ3-η1-C≡CC6H4C≡C–4)M3(μ-dppm)3](BF4)4 [39]. The electronic absorption spectra of these complexes are dominated by the high-energy absorption of the diphosphine and alkynyl intra-ligand π–π* transitions. Low-energy absorptions at ca. 324−416 nm are noted which are assigned as predominantly LMCT [acetylide → Cu4/M3] character. Mixing of MC d–s state and IL π–π* character is also possible, particularly in the case of the hexanuclear species.
These d10 alkynyl complexes are found to be strong triplet emitters in the green to orange region upon photo-excitation. Blue shift in emission energies is observed on going from the hexanuclear Cu(I) complex to the Ag(I) analogues, supportive of the assignment of a mixed LMCT[C≡C → M3]/MC[d–s] character in their lowest-lying emissive state. In view of the unusually long lifetime of the emissive state and the low-lying π* orbital energy of the diynyl unit, possible involvement of a 3IL [π–π*] character could not be completely excluded.
3.2 Rhenium alkynyl complexes
In addition to d10 metal complexes, our group has also been interested in the studies of other transition metal alkynyl complexes. In particular, a series of neutral rhenium(I) alkynyl complexes, [Re(diimine)(CO)3C≡CR] (R = aryl, alkyl) have been synthesised in our laboratory (Fig. 5) [14]. The electronic absorption spectra of these rhenium(I) alkynyl complexes show low-energy absorption bands at ca. 391−440 nm, attributed to a MLCT [dπ(Re) → π*(diimine)] transition. Upon photo-excitation, these complexes show long-lived intense luminescence in the solid-state and in fluid solutions at room temperature. Their emission energies are found to be easily tuned by the systematic variations of the substituents on the alkynyl ligand and the diimine ligand (Table 1) [14]. In general, a more electron-donating alkynyl ligand would give a lower energy MLCT emission and this is consistent with the σ- and π-donating ability of the alkynyl moieties. Thus, the long-lived emission for this class of complexes is assigned as originating from a dπ(Re) → π*(diimine) MLCT triplet state.
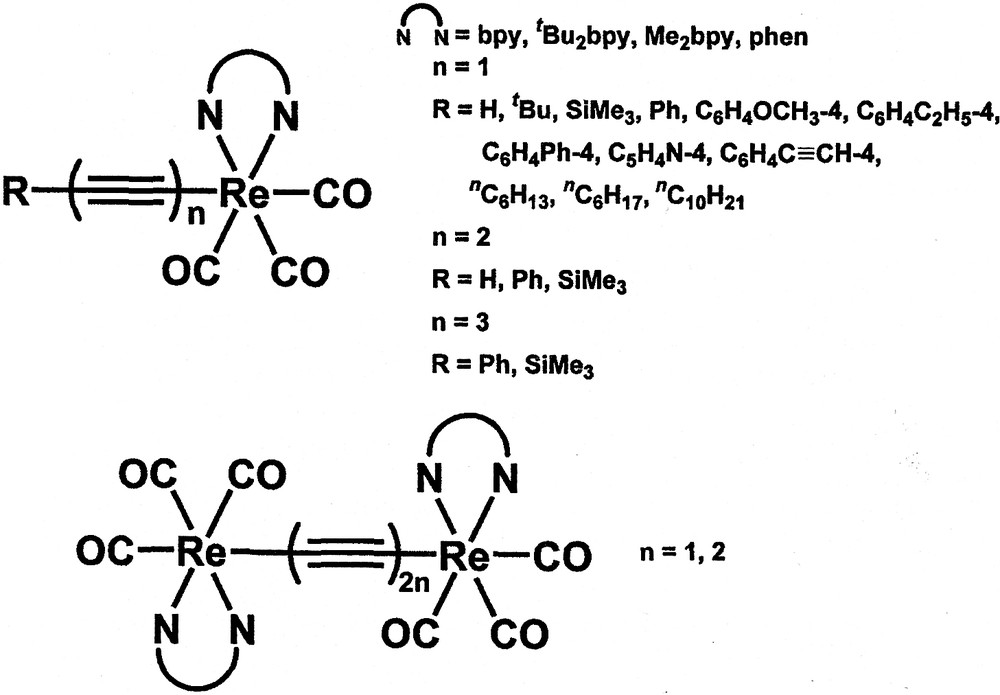
Mono- and dinuclear rhenium(I) diimine alkynyl complexes.
Electronic absorption and emission data for selected [Re(diimine)(CO)3C≡CR] illustrating the dependence of emission energies on the substituent on the alkynyl ligand
| Complex | Absorptiona λ/nm (ɛ/dm3 mol−1 cm−1) | Medium (T/K) | Emission λem/nm (τo/μs) |
| [Re(bpy)(CO)3–C≡C–Ph] | 286 (37,250) | Solid (298) | 585 (0.19) |
| 298 sh (30,160) | Solid (77) | 570 | |
| 326 sh (10,430), 420 (3240) | CH2Cl2 (298) | 654 (< 0.1) | |
| [Re(bpy)(CO)3–C≡C–C6H4–Cl-4] | 284 (39,880) | Solid (298) | 580 (< 0.1) |
| 298 sh (35,380) | Solid (77) | 565 | |
| 324 sh (13,330), 420 (2710) | CH2Cl2 (298) | 650 (< 0.1) | |
| [Re(bpy)(CO)3–C≡C–C6H4–C8H17-4] | 278 (36,120), 298 sh (29,200) | Solid (298) | 597 (0.27) |
| 328 sh (9570), 424 (2430) | Solid (77) | 588 | |
| THF (298) | 668 (< 0.1) |
a In CH2Cl2 at 298 K.
In view of the synthetic versatility of this class of complexes towards the use of different alkynyl ligands and the growing interest in the study of metal-containing rod-like complexes, a series of mono- and di-rhenium rod-like complexes with conjugated oligoynyl ligands, [Re(diimine)(CO)3(C≡C)nR] and [(CO)3(diimine)Re(C≡C−C≡C)nRe-(diimine)(CO)3], have been synthesised (Fig. 5) [14,40–42]. The luminescence properties have been found to be influenced by the number of C≡C units in addition to the substituents on the diimine and oligoynyl ligands (Table 2) [40,41]. In general, the triynyl complexes are found to emit at higher energies than their respective diynyl and monoynyl analogues. An assignment of a 3MLCT[dπ(Re) → π*(C≡CR)] or a metal-perturbed 3IL[π → π*{(C≡C)nR}] origin is not compatible with such a trend. Rather a 3MLCT[dp(Re) → π*(diimine)] assignment is suggested. Such anomalous energy trend in the 3MLCT emission bands is suggested to arise from the decreased overlap integral between the dπ(Re) and the triynyl π(RC≡C−C≡C−C≡C) orbitals than that of dπ(Re) and the π(RC≡C−C≡C) and π(RC≡C) orbitals resulting from the delocalisation of electron density across the (C≡C)n unit. As a result, the dπ(Re) is stabilised to a larger extent for complexes with more C≡C units giving rise to a higher energy 3MLCT emission. Thus, an assignment of a 3MLCT [dπ(Re) → π*(diimine)] origin for the lowest lying emissive state for these complexes is favoured.
Electronic absorption and emission data for selected oligoynyl rhenium(I) diimine complexes of different carbon chain length
| Complex | Absorptiona λ/nm (ɛ/dm3 mol−1 cm−1) | Medium (T/K) | Emission λem/nm (τo/μs) |
| [Re(tBu2bpy)(CO)3–C≡C–Ph] | 296 sh (21,060) | Solid (298) | 600 (0.24) |
| 406 (3550) | Solid (77) | 585 | |
| THF (298) | 688 (0.20) | ||
| [Re(tBu2bpy)(CO)3–C≡C–C≡C–Ph] | 248 (52,260) | Solid (298) | 570 (<0.1) |
| 298 (48,570) | Solid (77) | 570 (1.90, 0.32)b | |
| 340 (14,610), 416 (3220) | THF (298) | 625 (<0.1) | |
| [Re(tBu2bpy)(CO)3–C≡C–C≡C–C≡C–Ph] | 260 (60,740) | Solid (298) | 568 (<0.1) |
| 282 (70,290) | Solid (77) | 588 (1.20, 0.11)b | |
| 298 sh (63,100) | THF (298) | 632 (<0.1) | |
| 352 (13,370) | |||
| 378 (13,570), 426 (4540) |
a In CH2Cl2 at 298 K.
b Double exponential decay.
3.3 Mixed-metal alkynyl complexes
With the homo-metallic alkynyl systems discussed in previous sections in hand, we are interested in the exploration of luminescent mixed-metal molecular rods and materials.
3.3.1 Rhenium(I)–copper(I) and –silver(I) mixed-metal alkynyl complexes
The rhenium(I)–copper(I) mixed-metal alkynyl complexes, [M3(μ-P∧P)3{μ3-η1-C≡C−C6H4−C≡C-p-Re(N∧N)(CO)3}2]+ and [M3(μ-P∧P)3{μ3-η1-C≡C−C≡C-p-Re(N∧N)(CO)3}2]+ [43,44], and decanuclear rhenium(I)–silver(I) alkynyl complexes, [Ag6(μ-dppm)4{μ3-C≡CC≡C−Re(N∧N)(CO)3}4](PF6)2 [45], were synthesised using a metalloligand approach (Fig. 6).
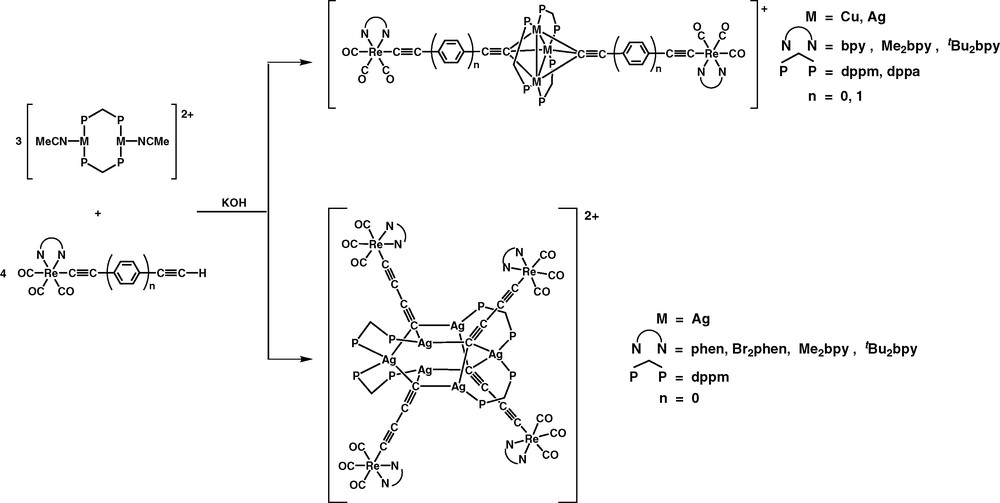
Synthesis of Re(I)–Cu(I) and –Ag(I) mixed-metal alkynil complexes via the metalloligand approach.
The complex cations of the pentanuclear Re(I)–Cu(I) and Re(I)–Ag(I) mixed-metal complexes consist of an isosceles triangle of the d10 metal atoms with a dppm ligand bridging each edge to form a roughly planar {M3P6} core, with two Re(I) butadiynyl metalloligands capping the M3 core in a μ3-η1 fashion. In contrast, the complex cations of the decanuclear complexes show a Ag6 core decorated with four Re(I) butadiynyl metalloligands, with each of the silver atom linked to two bridging diphosphine ligands and each butadiynyl group has one end coordinated to three silver atoms in a μ3-η1 bonding mode and the other end coordinated to a rhenium atom. The Ag6 core consists of two Ag3 triangular planes that are arranged in a nearly perpendicular fashion.
The electronic absorption spectra of these mixed-metal complexes show high-energy absorption bands typical of intraligand π–π* transitions of the diphosphines, alkynyl and diimine ligands. In addition, low-energy absorption bands at ca. 434−482 nm are also noted and the absorption energies of these bands are found to be sensitive to the nature of the diimine ligands, with the trend in line with the π-acceptor ability of the diimine ligands: Br2phen < phen < Me2bpy ≤ tBu2bpy, and the low-energy absorptions are assigned as MLCT [dπ(Re) → π*(N∧N)] transitions (Br2phen = 4,7-dibromo-1,10-phenanthroline, phen = 1,10-phenanthroline, Me2bpy = 4,4′-dimethyl-2,2′bipyridine, tBu2bpy = 4,4′-di-tert-butyl-2,2′-bipyridine). In general, slight red shifts are noted on going from the pentanuclear Re(I)–Ag(I) complexes to the corresponding Re(I)–Cu(I) analogues, in line with the poorer electron-donating ability of the AgI3 core than the CuI3 counterpart, which would render the Re(I) centre less electron-rich, leading to a larger cluster-perturbed MLCT energy gap [43–45].
Upon photo-excitation, these mixed-metal complexes are found to exhibit intense orange to red luminescence in the solid-state and in fluid solutions, which is at a lower energy region when compared to their corresponding precursor complexes. Similar to the electronic absorption spectroscopy, the emission energies are found to be dependent on the nature of the diimine ligand, diphosphine ligand, and the metal core. Degassed dichloromethane solutions of the complexes with different diimine ligands show emission energy dependence on the π* orbital energies of the diimine ligands. As an example, the trinuclear complex [Cu3(μ-dppm)3{μ3-η1-C≡CC≡C−Re(bpy)(CO)3}2]+ emits at 706 nm, while its tBu2bpy analogue emits at 678 nm. With reference to previous spectroscopic work on rhenium(I) diimine systems, the intense low-energy emissions are assigned as emanated from 3MLCT [dπ(Re) → π*(diimine)] excited state, probably mixed with some 3LLCT [π(C≡CC≡C) → π*(diimine)] character. A slight blue shift in emission energies is observed when changing the diphosphine ligand in [Cu3(μ-P∧P)3{μ3-η1-C≡CC≡C−Re(bpy)(CO)3}2]+ from P∧P = dppm (706 nm) to dppa (701 nm) and in [Cu3(μ-P∧P)3{μ3-η1-C≡CC≡C−Re(Me2bpy)(CO)3}2]+ from P∧P = dppm (688 nm) to dppa (677 nm). This is in accordance with the stronger electron-donating ability of dppm than dppa, which would render the Cu(I) alkynyl moieties more electron-rich and in turn render the cluster-modified Re(I) centre more electron-rich, raising the energy of the dπ(Re) orbitals and finally resulted in a lower energy 3MLCT emission. In view of the perturbation of emission energies by changing the diphosphine ligand and the d10 metal centre, the lowest-lying emissive state in these mixed-metal complexes are best described as having predominantly 3MLCT [dπ(Re) → π*(diimine)] character, probably mixed with some 3LLCT[π(C≡CC≡C) → π*(diimine)] character that has been perturbed by the d10 metal core.
In view of the versatile bonding motif exhibited by the alkynyl group, we are also interested in the construction of mixed-metal alkynyl complexes via the π-bonding mode of the alkynyl group. A series of mixed-metal η2-alkynyl-coordinated complexes of rhenium(I)–copper(I) and –silver(I), [{η2-Re(CO)3(bpy)(C≡CPh)}2M]+ (M = Cu, Ag), have been successfully isolated in our laboratory (Table 3) [46]. The metalloligand [Re(CO)3(bpy)C≡CPh] react with the corresponding d10 metal complexes [M(MeCN)4]PF6 (M = Cu, Ag) in THF at room temperature in an inert atmosphere of nitrogen to give the π-bonded species as yellow crystals after recrystallisation. Low-energy absorptions at ca. 396 nm are noted in the electronic absorption spectra of the complexes, which are assignable as a MLCT [dπ(Re) → π*(bpy)] transition. The blue shift of the lowest-energy absorption band of the π-bonded species [M = Ag (600 nm), Cu (590 nm)] compared to their precursor complex [Re(CO)3(bpy)C≡CPh] (654 nm) is a consequence of the weaker π-donating ability of the alkynyl ligand upon π-coordination to the d10 metal ions which would stabilise the dπ(Re) orbitals to a larger extent. Similar trends have also been observed in the emission of the mixed-metal complexes, and the emission is assigned accordingly as derived from states of a 3MLCT [dπ(Re) → π*(bpy)] character [46].
Electronic absorption and emission data for mixed-metal rhenium(I)–copper(I) and –silver(I) alkynyl complexes
| Complex | Absorptiona λ/nm (ɛ/dm3 mol−1 cm−1) | Medium (T/K) | Emission λem/nm (τo/μs) |
| [Re(bpy)(CO)3–C≡C–Ph] | 286 (37,250) | Solid (298) | 585 (0.19) |
| 298 sh (30,160) | Solid (77) | 570 | |
| 326 sh (10,430), 420 (3240) | CH2Cl2 (298) | 654 (<0.1) | |
| [{η2-Re(CO)3(bpy)(C≡CPh)}2Ag]+ | 250 (26,890), 292 (28,240) | Solid (298) | 535 (<0.1) |
| 396 (5930) | Solid (77) | 533 | |
| CH2Cl2 (298) | 600 (0.16) | ||
| [{η2-Re(CO)3(bpy)(C≡CPh)}2Cu]+ | 246 (28,160), 288 (29,000) | Solid (298) | 550 (0.35) |
| 396 (6050) | Solid (77) | 540 | |
| CH2Cl2 (298) | 590 (0.18) |
a In CH2Cl2 at 298 K.
3.3.2 Rhenium(I)–iron mixed-metal alkynyl complexes
Apart from the rhenium(I) mixed-metal complexes with coinage metals, we are also interested in the design and synthesis of mixed-metal species of different metal centres. In the collaboration between our group and the groups of Lapinte and Halet, an electro-switchable luminescent complex, [(CO)3(bpy)Re(C≡CC6H4C≡C)Fe(C5Me5)(dppe)] was synthesised [47]. It is envisioned that heterometallic systems with redox-active metal centres would provide a promising and versatile strategy for the design of electro-switchable molecular materials.
3.4 Molecular platinum rods
The development of luminescent materials with 3MLCT emission has aroused much attention in recent years. Apart from the rhenium(I) alkynyl systems, our group has designed and synthesised a series of platinum-containing carbon-rich materials, [Pt(trpy)C≡CR]+, which also emit from a 3MLCT manifold [48]. With the on-going interest in the chemistry of carbon-rich metal-containing systems, in particular those of metal-containing polynes, we have directed one of our goals towards the study of luminescent platinum(II) terpyridyl-capped carbon-rich molecular rods. Recently, a series of luminescent molecular platinum rods, [Pt(tBu3trpy)(C≡C)nPt(tBu3trpy)](X)2 (n = 1, 2, X = OTf; n = 4, X = PF6) have been successfully synthesised [49].
The platinum atoms of the complex cations of these molecular platinum rods adopt an essentially square-planar geometry with slight distortion due to the coordination constraints imposed by the terpyridyl ligand. The two platinum(II) terpyridyl ends are connected by the alkynyl chain in a linear [Pt](C≡C)n[Pt] fashion with n = 1, 2 and 4. The dimension of the carbon-rich platinum rods extends from molecular scale [n = 1 (5.16 Å), 2 (7.71 Å)] to nanoscale [n = 3 (12.83 Å)].
The electronic absorption spectra of these platinum-containing carbon-rich materials show intense absorption bands at about 308−342 and 428−522 nm in acetonitrile at 298 K. With reference to previous spectroscopic studies on the related mononuclear [Pt(tBu3trpy)C≡CC≡CH](OTf) [50] and related complexes [48], the high-energy structured absorption bands at ca. 308−342 nm are assigned as intra-ligand (IL) π → π*[(C≡C)n] and π → π*(tBu3trpy) transitions of the alkynyl bridge and the terpyridine ligand, while the low-energy band at ca. 428−522 nm is ascribed to a MLCT[dπ(Pt) → π*(tBu3trpy)] transition, probably mixed with some ligand-to-ligand charge transfer LLCT[π(C≡C)n → π*(tBu3trpy)] character. A blue shift of the lowest-energy absorption band in these complexes is observed with an increasing number of C≡C units [n = 1 (484−522 nm), 2 (436−466 nm), 4 (428−452 nm)]. This anomalous observation is likely to be a consequence of the greater stabilisation of the dπ(Pt) orbital upon increasing the length of the conjugated carbon chain. This can be accounted by the π-donor ability exhibited by the alkynyl chain, which gives rise to a filled–filled pπ–dπ overlap, and the π-acceptor ability of the alkynyl chain, which results in a dπ–π* interaction. A similar argument has also been suggested to rationalise the trend observed in the related rhenium(I) alkynyl system [14].
Photo-excitation of the platinum molecular rods in the solid-state and in fluid solutions give intense luminescence at ca. 550−625 nm. The occurrence of the emission at similar energies to that found in the related mononuclear complex [Pt(tBu3trpy)C≡CC≡CH](OTf) [50] suggested that the origin of the luminescence is derived from states of predominantly 3MLCT[dπ(Pt) → π*(tBu3trpy)] character, probably mixed with some intra-ligand 3IL[π → π*(C≡C)n], and 3LLCT[π(C≡C)n → π*(tBu3trpy)] character. The emission spectra of the monoynyl and diynyl complexes in glass at 77 K exhibit vibronically structured bands with vibrational progressional spacings of about 1250 cm−1, which is typical of the ν(C···C) and ν(C···N) stretching modes of the terpyridine ligand, while the emission spectrum of the tetraynyl complex in glass at 77 K show vibronically structured bands with vibrational progressional spacings of ca. 2100 cm−1, typical of the ν(C≡C) stretch in the ground state. These observations are indicative of the involvement of the terpyridine ligand and alkynyl bridge in the excited state for these complexes. For reasons discussed for the electronic absorption spectroscopy, a blue shift of the emission energies was also observed on going from the monoynyl complex to the diynyl complex in butyronitrile glass at 77 K. It is likely that the emission of the tetraynyl complex would involve an increased contribution from the 3IL[π → π*(C≡C)n] excited states, which account for the observed lowest emission energy and vibrational progressional spacings of ca. 2100 cm−1. Therefore, the emission for these complexes could best be described as an admixture of 3MLCT[dπ(Pt) → π*(tBu3trpy)], 3LLCT[π(C≡C)n → π*(tBu3trpy)], and 3IL[π(C≡C) → π*(tBu3trpy)] character, with an increase in the 3IL contribution upon increasing the π-conjugation of the alkynyl bridge.
4 Metal-based luminescent sensors
The polymorphic behaviour of square-planar platinum(II) polypyridyl complexes has attracted attention in recent years [15–19]. The colour changes in their solid states are dependent on the extent of metal···metal interactions and π–π stacking of the polypyridyl ligands. These have prompted us to investigate any polymorphic behaviour exhibited by the platinum(II) terpyridyl alkynyl complexes and their potential applications as metal-based luminescent probes and reporters. The platinum(II) terpyridyl butadiynyl complex, [Pt(trpy)C≡CC≡CH]OTf, was found to exist in two forms, a dark green form and a red form with different crystal packing arrangements [50]. The platinum atoms in the dark green form are almost superimposable to give a nearly perfect linear [Pt]n chain when viewed along the Pt–Pt axis, with the platinum atoms equally spaced and with short intermolecular distance of ca. 3.388 Å. The individual molecules are rotated with respect to their neighbours, with a partial stacking of the aromatic terpyridyl units. On the other hand, the red form shows dimeric structure with alternating Pt···Pt distances of 3.394 and 3.648 Å (Table 4). The ‘short’ and ‘long’ Pt···Pt bonds exhibit a zigzag arrangement. On the contrary, a related complex, [Pt(tBu3trpy)C≡CC≡CH]OTf, exists as discrete monomeric units with no evidence of short Pt···Pt contacts, probably a consequence of the steric demand of the bulky tert-butyl groups on the terpyridine ligand.
Solid-state electronic absorption, emission and structural data for [Pt(trpy)C≡C–C≡CH]OTf at 298 K
| Crystal form | Intermolecular Pt···Pt distances/Å | Absorption λ/nm | Emission λem/nm |
| Red | 3.394, 3.648 | 510 | 680 |
| Dark-green | 3.388 | 685 | −a |
a Beyond the detectable range of the instrument.
Unlike the solid-state absorption in which the green form absorbs at 685 nm and the red form at 510 nm, the electronic absorption spectra of the two forms of [Pt(trpy)C≡CC≡CH]OTf in acetonitrile or acetone are identical and are dominated by high-energy intra-ligand absorptions and a low-energy MLCT absorption at 416 nm, typical of monomeric platinum(II) terpyridyl complexes. Upon increasing the diethyl ether content in solution, the colour of the solution changed dramatically from yellow to green to blue, with a new absorption band formed at 615 nm, concomitant with a drop in absorbance at 416 nm, and a tremendous emission enhancement at 785 nm in the near-infrared region was observed. In contrast, there is no observable colour change in the related [Pt(tBu3trpy)C≡CC≡CH]OTf in various solvent mixtures. The drastic colour changes displayed by the solutions of [Pt(trpy)C≡CC≡CH]OTf are believed to be associated with the formation of aggregates in solution as a result of the reduced solvation upon increasing the diethyl ether composition. The tremendous increase in emission intensity at 785 nm upon increasing the diethyl ether composition is also attributed to aggregate formation through metal···metal interactions and π–π stacking. Assignments of the absorption band at ca. 615 nm to a metal–metal-to-ligand charge transfer (MMLCT) transition and the phosphorescence at ca. 785 nm as derived from states of a 3MMLCT character have been made. This kind of solvent-induced aggregation may be regarded as a special type of solvatochromism. These carbon-rich complexes may serve as potential probes and reporters of environmental changes and may find potential applications in materials science and sensor technology.
Acknowledgements
Financial support from the Research Grants Council of the Hong Kong SAR, China (HKU 7123/00P) and The University of Hong Kong Foundation for Educational Development and Research Limited are gratefully acknowledged.