

1 Introduction
Manganese, a group–7 metal, possesses possibly the largest number of easily accessible oxidation states. Manganese is extremely essential trace element for man, but when inhaled in mines it can trigger Parkinson like symptoms [1]. Also an organometallic manganese compound, used in gasoline as a substitute of tetraethyl lead impairs man's neurological health [2] even on slight inhalation. Contextually, hydroxamic acids as ligands are important because they are well known for their siderophoric roles [3], often being produced by bacteria and algae to sequester and facilitate the transport of metal ion nutrients into the cell [4]. One such siderephore (trade name ‘desferal’) is used as a drug for the treatment of iron overload in thalassaemia [5]. Based on this as well as other medicinal uses as aspirin analog, nitric oxide donor, inhibitors of prostaglandin H synthase, peroxidases and ureases as well as the fact that a hydroxamic acid (trade name Marimastat) is going to appear as anticancer drug [5] very soon, a detailed study of Mn-hydroxamic acid reaction is more than warranted. Strikingly, manganese-hydroxamate chemistry is still unknown and a few years ago we made the inaugural report [6] of manganese hydroximate chemistry where capability of primary hydroxamic acid to form stable hydroximate-complexes of Mn(III) and Mn(IV) from Mn(II) starting material in aqueous–aerobic medium, as against formation of only polymeric Mn(II) hydroxamate complexes with secondary hydroxamic acids, under the same experimental conditions, was attributed mainly to electronic reasons [6]. We have now noted that BPHAH (see Scheme 1; for the authenticity that the ligands have their nomenclature as hydroxamic acids and not hydroxylamines [7a] see [7b,8–12]) on reaction with Mn(II) yield polymeric [MnII(BPHA)2]n, as we observed earlier, but an ortho substitution in the N-phenyl rings of the same ligand afford discrete mononuclear Mn(III) complexes, namely [MnIII(BOTHA)3] (2) (BOTHAH = N-benzoyl N-ortho-tolyl hydroxamic acid (A2)) and [MnIII(BOCPHA)3] (3) (BOCPHAH = N-benzoyl N-orthochloro phenyl hydroxamic acid (A3)), maybe due to the steric hindrance which prevents the polymerization, because instead of ortho, when the same substituents occur in meta (A4, A5) or para (A6, A7) position of the same N-phenyl ring of the ligand framework, a light yellow and highly polymeric (insoluble in water as well as in the entire range of organic solvents) materials having empirical formula: [Mn(hydroxamate)2]n (4–7) is precipitated. Interestingly, if the B type ligand (Scheme 1) N-phenyl N-cinnamoyl hydroxamic acid (CPHAH, B1) is used, also a polymeric Mn(II) complex of the type [Mn(CPHA)2]n′ (8) (n′ may not be equal to n) is obtained, but it has a light brown color. Unfortunately, it was not possible for us to obtain diffractable crystals of the soluble complexes (2) and (3). Under this situation we report here the synthesis, analytical, spectroscopic, magnetochemical, EPR, electrospray mass and electrochemical characterization of the isolated complexes along with the homogeneous catalytic properties of 2 and 3 in the epoxidation of olefenic compounds using H2O2 as terminal oxidant. Notably, epoxidation of olefins is a very outstanding transformation in organic synthesis, since the epoxy compounds are widely used as such or for manufacturing wide variety high demand commodity chemicals like polyurethans, unsaturated resins, surfactants and other products [13]. Hydrogenperoxide is probably the best terminal oxidant after dioxygen with respect to environmental and economic considerations. Indeed in certain circumstances it is better than dioxygen insofar as O2–organic mixtures spontaneously ignite. However, manganese-complex catalyzed epoxidation was pioneered by Zhang and Jacobsen [14].

Structural formula and abbreviation of the ligands used.
2 Experimental section
2.1 Materials
Ethanol, dichloromethane, acetonitrile, benzoyl chloride, benzene and zinc powder were of analytical grade and obtained from E. Merck (India). Extra pure variety of sodium hydrogen carbonate and o-, m-, and p-nitrotoluene were obtained from Sarabhai-Merck Chemicals (Mumbai). Cinnamic acid, phosphorous pentoxide and o-chloro nitrotoluene were of extrapure variety of LOBA Chemie (India). Thionyl chloride (A.R.) was obtained from spectrochem (India), sodium acetate from S.D. fine chemicals and Mn(OAc)2·4 H2O from Qualigen fine chemicals, India. Tetraethyl ammonium perchlorate (TEAP) for electrochemical work was prepared using known method [15]. The hydroxamic acids (though named as hydroxylamines) used here were synthesized using the recipe reported earlier [7]. The GC standards viz. the authentic epoxides were obtained from Lancaster Synthesis Chemical (Research grade) Chennai, India, HPLC grade (acetonitrile and diethyl ether) was obtained from Sisco Research Laboratories (SRL), Mumbai.
2.2 Physical measurements
The IR spectra were recorded as KBr pellets on a Perkin–Elmer 597 IR spectrophotometer and Electronic spectra were obtained from a Hitachi Model U-3410 UV–Vis–NIR spectrophotometer. The magnetic susceptibilities were obtained from a vibrating sample magnetometer by Gouy Method utilizing Hg[Co(NCS)4] as a standard. Elemental analysis (C, H, N) was performed micro-analytically using a Perkin–Elmer 240C elemental analyzer and manganese was estimated by the literature method [16]. Molecular weights of 2 and 3 were measured by a Knauer (Berlin) vapor pressure osmometer using benzyl as standard. Electronspray mass spectra were recorded on a Micromass Auattro II triple quadropole mass spectrometer (CDRI, Lucknow). Cyclic voltammetric measurements were made with the use of a PAR model 378-1 electrochemical system. X-Band EPR spectra of the powdered samples were recorded on a Varian E-112 spectrometer at room temperature. The spectra were calibrated with diphenyl picrylhydrazyl (dpph) (g = 2.0037) radical. The Chromatographic analysis was performed using a Agilent model 6890 N Gas Chromatograph using HP-1 and INNOWAX Capillary Column in FID mode with dinitrogen as carrier gas maintaining the oven temperature at 40 °C for 5 min and then programmed to 160 °C at 10 °C min–1.
2.3 Preparation of the complexes
2.3.1 Bis (N-benzoyl N-phenyl hydroxamato) manganese (II) (1)
Mn(OAc)2·4 H2O (0.15 g; 0.61 mmol) was dissolved in minimum volume (5 ml) of water and to it was added dropwise and with stirring an ethanol solution (10 ml) of BPHAH (0.39 g; 1.8 mmol). A light yellow solid was separated and the stirring was continued for a further 1 h when the solid was filtered off and washed with ethanol and diethyl ether. The same was dried and collected. Yield 67% (0.76 mmol). The substance was insoluble in water as well as in organic solvents. Anal. Calc: for C26H20N2O4Mn; C, 65.13; H, 4.41; N, 5.86; Mn, 11.5. Found: C, 65.09; H, 4.14; N, 5.98; Mn, 11.2%. IR (KBr disc, cm–1): 1600(s) and 1570(s) [ν(C=O)] [17], 1510(m) [ν(C–N)] [18], 1440(m), 1290(m), 1160(m) [ν(N–O)] [18], 1080(w), 1040(w), 1020(s), 1000(m), 930(s), 790(m), 770(s), 740(m), 720(m), 690(s), 670(m), 590(w), 440(w), 390(w), 320(w).
2.3.2 Tris (N-benzoyl N-O-tolyl hydroxamato) manganese (III) (2)
Mn(OAc)2·4 H2O (0.15 g; 0.61 mmol) was dissolved in minimum volume (5 ml) of water and to it was added dropwise and with stirring an ethanol solution (20 ml) of BOTHAH (0.55 g; 2.45 mmol). A deep green solid separated and the stirring was continued for a further 3 h when the solid was filtered off and washed with water and diethyl ether. The same was dried in vacuo. Yield 65% (0.52 mmol). The substance was soluble in most organic solvents and the product was recrystalized from acetonitrile. Anal. Calc: for C42H36N3O6Mn; C, 68.76; H, 4.91; N, 5.73; Mn, 7.32%, M.W. 733.42 Found: C, 68.40; H, 5.71; N, 5.47; Mn, 7.50%, M.W. 715. IR (KBr disc, cm–1) 1550(s) [ν(C=O)], 1510(s) [ν(C–N)], 1440(m), 1160(m) [ν(N–O)], 1120(w), 1080(w), 1020(s), 920(s), 770(s), 700(s), 660(m), 620(m), 540(w), 520(w), 450(w). UV–Vis (nm, molar absorption as M–1 cm–1 in parentheses): 685 (292), 476 (575), 270 (25090).
2.3.3 Tris (N-benzoyl N-ortho-chlorophenyl hydroxamato) manganese (III) (3)
The same method as that of 2 was followed using BOCPHAH instead of BOTHAH. Yield 68% (0.54 mmol). The substance was soluble in most organic solvents. Anal. Calc: for C39H27N3O6Cl3 Mn; C, 58.91; H, 3.4; N, 5.30; Mn, 7.12; M.W, 794.96. Found: C, 58.10; H, 3.88; N, 4.98; Mn, 6.92%; M.W, 765. IR (KBr disc, cm–1): 1590(m) and 1550(s) [ν(C=O)], 1500(m) [ν(C–N)], 1430(s), 1160(m) [ν(N–O)], 1080(s), 1010(s), 930(s), 920(s), 770(s), 690(s), 680(m), 650(s), 550(w), 450(w). UV–Vis (nm, molar absorption as M–1 cm–1 parentheses): 701 (450), 484 (960), 271 (27870).
Notably, for 2 and 3 a ligand to metal mole ratio of at least 4:1 is necessary to get the maximum yield.
2.3.4 Bis (N-benzoyl N-meta-tolyl hydroxamato) manganese (II) (4) and bis (N-benzoyl N-meta-chloro phenyl hydroxamato) manganese (II) (5)
Mn(OAc)2·4 H2O (0.15 g; 0.61 mmol) was dissolved in minimum volume (5 ml) of water and to it was added dropwise and with stirring an ethanol solution (20 ml) of BMTHAH (0.42 g; 1.8 mmol) or BMCPHAH (0.45 g; 1.8 mmol), respectively. Light yellow solid was separated in each case and the stirring was continued for a further 1 h. The solids were filtered off and washed with ethanol and diethyl ether. The same were dried and collected. Yield 65% (0.73 mmol) and 62% (0.73 mmol), respectively. The substances were insoluble in water as well as in organic solvents. Anal. Calc: for C28H24N2O4Mn; C, 66.5; H, 4.92; N, 5.27; Mn, 10.57; Found (for 4): C, 66.27; H, 4.73; N, 5.52; Mn, 10.85%. Calc: for C26H18N2O4Cl2Mn; C, 57.21; H, 3.19; N, 4.86; Mn, 10.24; Found (for 5): C, 56.93; H, 3.28; N, 5.11; Mn, 10.04%. IR (KBr disc, cm–1) (for 4): 1600(s) and 1560(s) [ν(C=O)], 1510(m), 1460(m) [ν(C–N)], 1440(w), 1160(m) [ν(N–O)], 1060(m), 950(s), 840(m), 780(w), 710(m), 700(s), 690(w), 660(m), 430(w). IR (KBr disc, cm–1; for 5): 1570(s) and 1550(s) [ν(C=O)], 1520(m), 1450(m) [ν(C–N)], 1350(s), 1260(w), 1160(m) [ν(N–O)], 1060(s) 1030(m), 930(w), 860(s), 780(m), 770(s), 720(m), 700(s), 680(m), 660(m), 620(m), 430(w).
2.3.5 Bis (N-benzoyl N-para-tolyl hydroxamato) manganese (II) (6) and bis (N-benzoyl N-para-chloro-phenyl hydroxamato) manganese (II) (7)
Mn(OAc)2·4 H2O (0.15 g; 0.61 mmol) was dissolved in minimum volume (5 ml) of water and to it was added dropwise and with stirring an ethanol solution (20 ml) of BPTHAH (0.42 g, 1.8 mmol) or BPCPHAH (0.46 g; 1.8 mmol), respectively. Light yellow solid separated in each case and the stirring was continued for a further 3 h. The solids were filtered off and washed with ethanol and diethyl ether. The same were dried and collected. Yield 65% (0.73 mmol) and 63% (0.71 mmol). The substances were insoluble in water as well as in organic solvents. Anal. Calc: for C28H24N2O4Mn; C, 66.5; H, 4.92; N, 5.27; Mn, 10.57%. Found (for 6) C, 66.37; H, 4.92; N, 5.52; Mn, 10.58%. Anal. Calc: for C26H18N2O4Cl2Mn; C, 57.21; H, 3.19; N, 4.86; Mn, 10.24%. Found (for 7) C, 56.86; H, 3.38; N, 5.12; Mn, 10.06%. IR (KBr disc, cm–1) (for 6): 1590(s) and 1560(s) [ν(C=O)], 1520(m), 1450(m) [ν(C–N)], 1410(w), 1160(m) [ν(N–O)], 1110(w), 1030(m), 930(s), 860(m), 820(s), 780(m), 700(s), 690(s), 620(w), 580(m), 520 (w), 470(w), 440(m), 360(w). IR (KBr disc, cm–1) (for 7): 1590(s) and 1550(s) [ν(C=O)], 1520(m), 1460(m) [ν(C–N)], 1320(m), 1160(m) [ν(N–O)], 1050(s), 1030(m), 930(m), 870(m), 780(m), 760(s), 720(m), 680(m), 650(s), 620(m), 510(w), 430(w).
2.3.6 Bis (N-Cinnamoyl N-phenyl hydroxamato) manganese (II) (8)
Mn(OAc)2·4 H2O (0.15 g; 0.61 mmol) was dissolved in minimum volume (5 ml) of water and to it was added dropwise and with stirring an ethanol solution (20 ml) of CPHAH (0.45 g; 1.8 mmol). A light brown solid separated and the stirring was continued for a further 1 h. The solid was filtered off and washed with ethanol and diethyl ether. The same was dried and collected. Yield 63%. The substance was insoluble in water as well as in organic solvents. Anal. Calc: for C30H24N2O4 Mn; C, 67.39; H, 4.26; N, 5.23; Mn, 10.38%. Found: C, 67.80; H, 4.52; N, 5.27; Mn, 10.36%. IR (KBr disc, cm–1): 1620(m) [ν(C=C), aliphatic] [19], 1560(s) [ν(C=O)], 1480(m) [ν(C–N)], 1440(m), 1220(m) [ν(N–O)], 1030(s), 1000(s), 980(w), 960(m), 860(w), 840(w), 780(m), 680(s), 580(w), 500(w), 470(w), 360(w).
3 Procedure for catalytic oxidation
0.5 × 10–5 mol catalysts (complex 2 or 3) was dissolved separately in acetonitrile (10 ml) in two different two neck (100 ml capacity) flat-bottomed flask to one neck of which was inserted a reflux condenser (to check evaporation loss) and to the other was fitted a stout septum. Substrates (0.5 × 10–2 mol), NaHCO3 (25 mol% of the substrates) and hydrogen peroxide (30% V/V; 3.0 ml, 0.02 mol) were added on each of the flask on stirring and same was continued (for the period of stirring see Tables 1 and 2) at room temperature (25 °C). Another 7.0 ml H2O2 (0.06 mol) was injected to the stirred solution, periodically (for the reaction which were completed within half an hour or so, addition of the entire 7.0 ml of H2O2 was not necessary). When the reaction was over, the respective solutions were taken in a separating funnel and repeatedly extracted with diethyl ether. The extract was then concentrated to 0.2 ml, from which 1 μl was withdrawn in a gas syringe and injected to the G.C. port. The retention times of the products were calibrated using authentic commercial standards. Product and catalyst isolation was not very difficult. The ether extract quantitatively takes out the substrate and the product. In case of 99% of product formation with respect to the substrate a preparative TLC was sufficient to make the product as pure as possible. For lesser conversion a recourse to the column chromatographic method was taken. The residue after ether extraction was rota-evaporated to expel the remaining organic solvent when the catalyst separated. Almost 0.1 × 10–5 mol catalyst could be recovered and the same was found to be still capable in inducing 'life' to the next batch of the reaction.
4 Results and discussion
Complexes 1, 4, 5, 6, 7 and 8 are insoluble in almost all solvents indicating that the compounds having the empirical formula Mn(BPHA)2, Mn(BMTHA)2, Mn(BMCPHA)2, Mn(BPTHA)2, Mn(BPCPHA)2 and Mn(CPHA)2 systems are highly polymeric. Complexes 2 and 3 are readily soluble in organic solvents viz., ethanol, methanol, acetone, acetonitrile, dichloromethane, benzene and chloroform and the solutions are non-electrolyte. From the vapor pressure osmometric data (see Section 2) in acetonitrile the molecular weights of 2 and 3, were assessed, which are nicely comparable to the calculated value. The ease of oxidation of Mn(II) aerially to Mn(III) or even to Mn(IV) is well documented [6,19]. In this connection two reports from Refs. [20,21] may be cited which give evidences that the degree of polymerization in [M(O-HA)2]x (O-HA = o-substituted hydroxamic acids) ligated with Ni(II) and Cu(II) , is much less than that of the complexes [M{M-/P-(HA)2}]x′ of same metal ions, that is, x << x′. Our results show that in the cases of o-substituted hydroxamic acid the steric hindrance restricted the polymerization reaction and in the mean time Mn(II) becomes oxidized to Mn(III) and then [MnIII(hydroxamate)3] can be isolated. This is a very interesting example where steric factor controls the oxidation state of metal–ligand complexes.
4.1 Magnetic susceptibility and EPR spectra
The room temperature magnetic moment values of 1, 4, 5, 6, 7 and 8 complexes are 5.89, 5.90, 5.92, 5.90, 5.88 BM, respectively, per molecular unit. This indicates that there does not exist any direct or super exchange of five unpaired spins of individual Mn2+ center at the ambient condition. The magnetic moment values of 2 and 3 are 4.88 and 4.83 BM, respectively, at 298 K which indicates that both are the Mn(III) complexes. As expected, 2 and 3 are EPR silent (Mn(III), d4) and the polymeric Mn(II) complexes, 1, 4, 5, 6, 7 and 8 are EPR active. Room temperature EPR spectrum of polycrystalline 8 displays (Fig. 1) most of the features of a typical rhombic Mn(II) spectrum. The g-tensors, viz. <g>1, <g>2 and <g>3 are 2.07, 2.01 and 1.98, respectively <g>av being 2.02 and the <A>isoMn = 109 G, with all the six Mn2+ hyperfine lines being resolved but one such <A> line is overlapped with a <g> line. Since all the ligand donor atoms are oxygen, the superhyperfines are absent (Fig. 1). Strikingly, all other Mn(II) polymers studied here show a broad isotropic signal with <g>iso = 2.04 (a representative spectrum is shown in the inset of Fig. 1). This difference in behavior of 8 with respect to other Mn(II) complexes is at present obscure, though it may be presumed that higher bulk of ligand B1 than those of others.
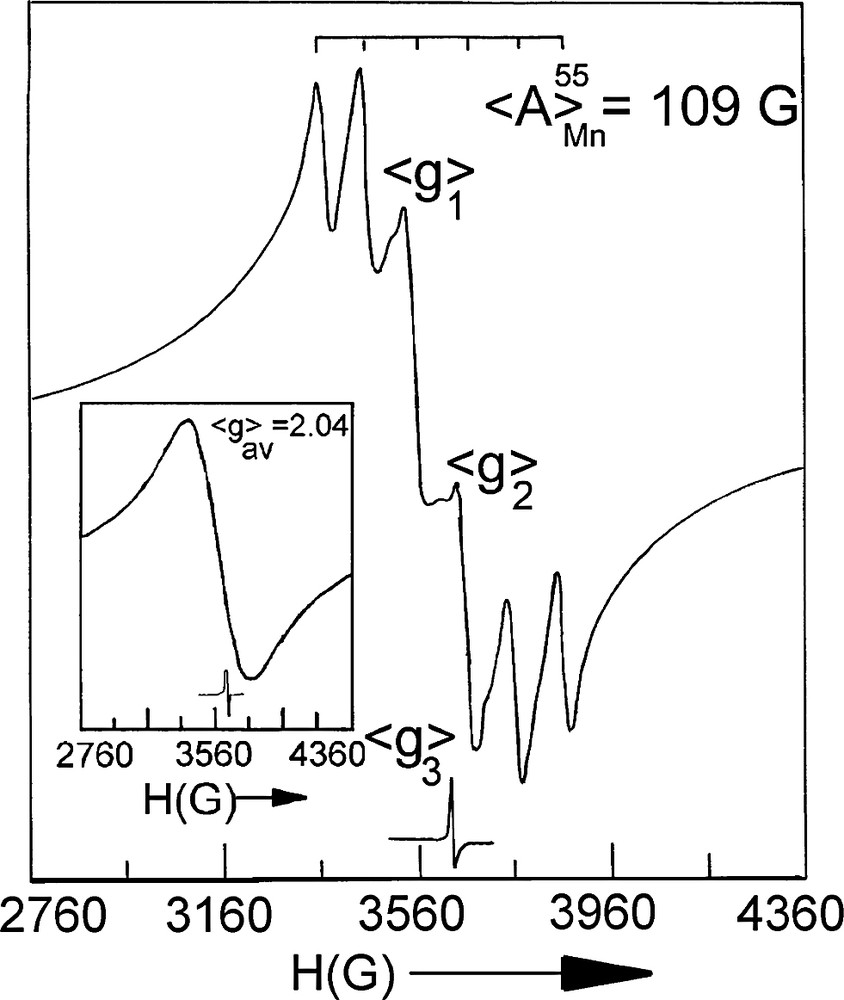
EPR spectrum of [MnII(CPHA)2]n′. In the inset is shown the isotropic spectrum of 1 as a representative case at 298 K.
4.2 IR and electronic spectroscopy
The ligands BPHAH(A1), BOTHAH(A2), BOCPHAH(A3), BMTHAH(A4), BMCPHAH(A5), BPTHAH(A6), BPCPHAH(A7), CPHAH(B1) exhibit two broad bands in the region 3120–2860 cm–1 which may be ascribed to ν(OH) vibration of N–OH function of the hydroxamic acids, the position of the band being drastically lowered due to strong intermolecular H-bonding of N–OH hydrogen and C=O oxygen. After complexation these broad bands totally disappeared indicating deprotonation of the –N−OH functional group on complexation. The intense ν(C=O) vibration at 1700 cm–1 in BPHAH shifts to 1600 and 1570 cm–1 [17] in complex 1 indicating a strong binding of two carbonyl oxygens (of 2 ligands) with manganese(II). The drainage of electron density from the oxygen centers in coordinated BPHA− to Mn(II) becomes so prominent that even a very strong and characteristic C−H out of plane deformation vibration of monosubstituted aromatic (benzene) ring [18] occurring at 740 cm–1 in A1 is also down shifted to 720 cm–1 in 1. The ligand A4–A7 behave similarly on metal complexation as is also expected. In B1 the ν(C=O) appears at 1590 cm–1 which shifts to 1560 cm–1 in 8 due to the same reasoning as put forward in BPHA− system. But the extent of the frequency lowering after complexation in 8 is not so pronounced as in the BPHA− as well as other hydroxamates studied here in so far as in CPHA– the C=O group is not attached to the aromatic ring, Moreover, the −HC=CH− residue existing in between benzene ring and C=O function in B1 shows the aliphatic ν(C=C) vibration at 1640 cm–1 as very sharp band which is also shifted after metal coordination of C=O group and appears at 1620 cm–1. In all the ligands a very prominent vibration appears at 1390 cm–1, which completely disappears in 2 and 5. It is also noteworthy that ν(N–O) of the free hydroxamic acid ligands occur at 1350−1190 cm–1 [18] region but after deprotonation and metal coordination via oxygen the same is shifted to lower wave number region viz. between 1220 and 1150 cm–1 [18] in all the complexes. The ν(C–N) vibration of the free ligands (A1–A7 and B1) appears near 1190 cm–1 as medium intensity band. Excepting in A2 and A3 and their Mn(III) complexes, the band at 1160 cm–1 remain unshifted. The extent of the shift of ν(C=O) vibration to lower wave numbers on complexation is greater in the case of the polymeric compounds than that in 2 and 3 may be due to the fact that C=O oxygen is more strongly bound with Mn(II) than with the Mn(III) center.
Solution spectra of only 2 and 3 could be taken due to solubility reason. The ligand A2 in acetonitrile medium shows a prominent band at 254 nm (7475 M–1 cm–1). Two other shoulders also appear from which Gaussian analysis brought out two bands with one at 345 and the other at 300 nm. Besides showing modified bands in this region, both 2 and 3 show two other bands at 685 and 476 nm, respectively. Those two d–d bands in this Jahn–Teller sensitive compounds (d4 manganese) should be assigned as 5B1 → 5B2 and 5B1 → 5A1 transitions, respectively [6]. The prominent U–V band of the ligand undergoes a positive shift appearing at 270 nm with ɛ almost three times higher than that of the pure A2 since 1 mole of complex 2 contains 3 moles of the ligand A2. The same ligand based π → π* transition in the case of A3 appears at 261 nm (ɛ = 8780 M–1 cm–1).
4.3 Electrochemical aspects
Cyclic voltammetry of [MnIII(BOTHA)3] (2) was carried out in CH3CN/TEAP/Pt and the voltamogram shows two oxidative cyclic responses at Ea 0.80 and 1.20 V, and a reductive response Ec at –0.65 V, vs. SCE which, however, is not shown in Fig. 2. The 0.80 V response is quasi reversible and its segmented form is shown as an inset in Fig. 2 with Ea = 0.83 V, Ec = 0.70 V and hence E0298 = 0.76 V and ΔEp = 130 mV and Ipa/Ipc = 1.03. This response can be assigned to the reaction as shown in Eq. (1). Under the same condition, E0298 of [η5C5H5] ⇌ [η5C5H5]+ at ΔEa = 0.80 V vs. SCE with ΔEp = 80 mV. For the second oxidative response, Ea = 1.19 V, Ec = 1.09 V and hence E0298 = 1.14 V, ΔEp = 100 mV (quasi-reversible), but Ipa/Ipc = 1.05. Hence, this oxidative response can be attributed to the couple as shown in Eq. (2).
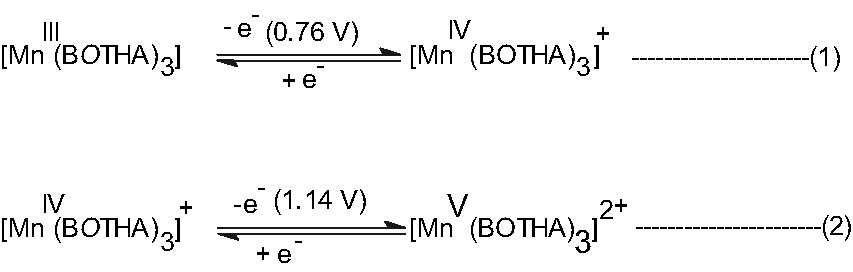

Cyclic voltammogram of [MnIII(BOTHA)3] in CH3CN/TEAP/Pt at a scan rate of 50 mV s–1, where Ec = cathodic peak potential, Ea = anodic peak potential, E0298 = (Ec + Ea)/2, ΔEp = Ea – Ec, Ipc = cathodic peak height, Ipa = anodic peak height.
4.4 Thermoanalytical studies
The thermograms for complexes 2 and 3 are shown in Fig. 3a, b, respectively, wherefrom it becomes apparent that the decomposition pattern of 2 and 3 significantly differs. Both the complexes show that the solids do not lose any lattice or coordinated water upto 275 °C for 2, 180 °C for 3. In 2 there appear two nearly horizontal intermediate regions (Fig. 3) and the most probable assignments of the molecular species formed giving these two breaks are shown in Scheme 2. The Scheme 2B also shows that the compound 3 decomposes at ca. 200 °C to the corresponding manganese oxides only. The reason for this difference in the mode of thermal decomposition in an inert atmosphere is, at present, obscure.
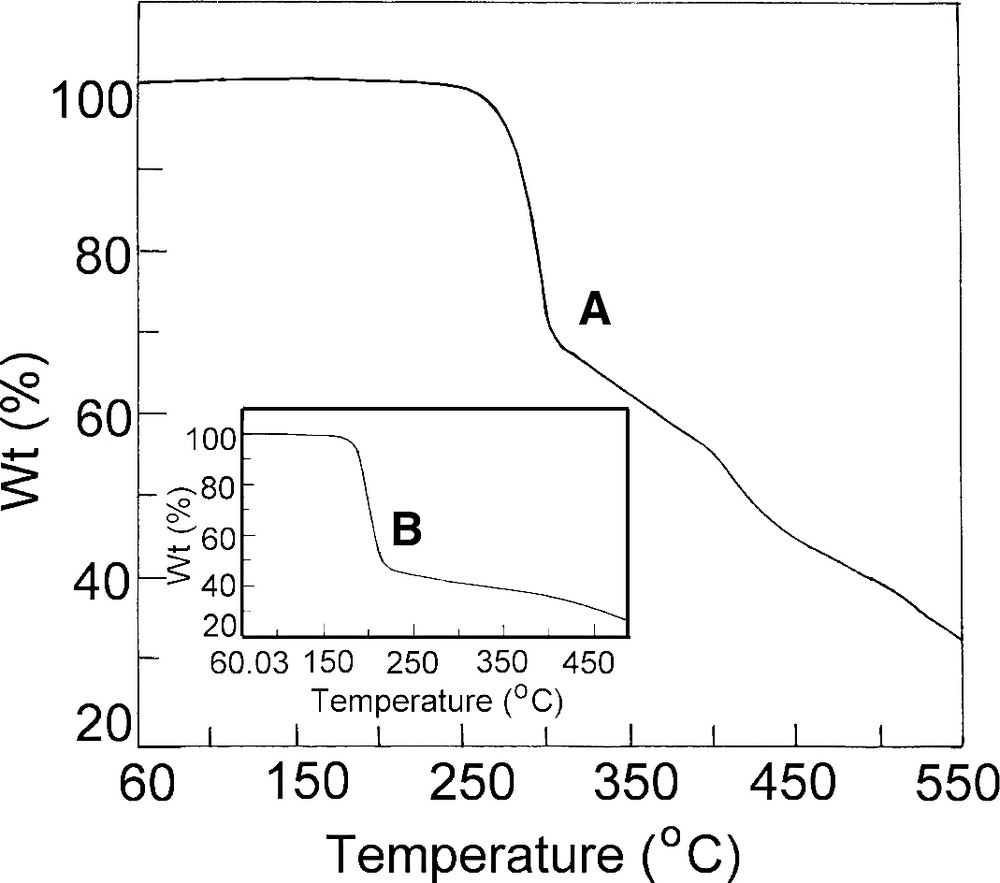
(A) Thermogravimetric analysis of complex [MnIII(BOTHA)3] and (B) [Mn(BOCPHA)3].
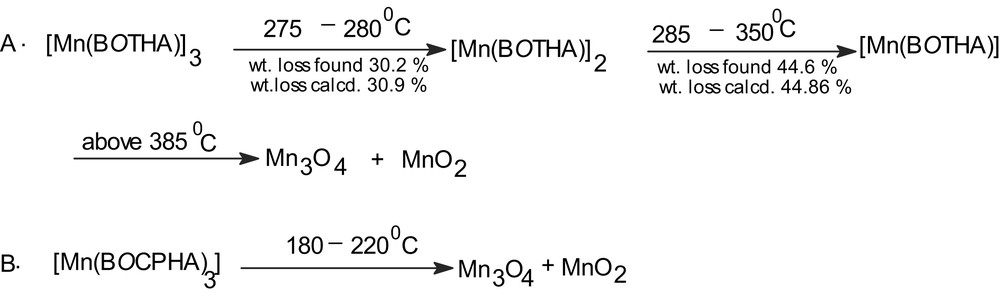
Mode of decomposition of 2 and 3 as revealed by TGA.
4.5 Electrospray mass spectrometry
The electrospray mass spectrometric analysis of 2 in the positive mode (Fig. 4a) shows that the molecular ion peak exists at m/z = 734 amu, but the peak height is very small. After that the sequence of fragmentation is shown in the Scheme 3a. The spectrum for 3 is also shown as Fig. 4b and sequence of fragmentation is given as Scheme 3b. So, all the physicochemical evidences including the electrospray mass spectral data support the formulae of the complexes, specially of 2 and 3.
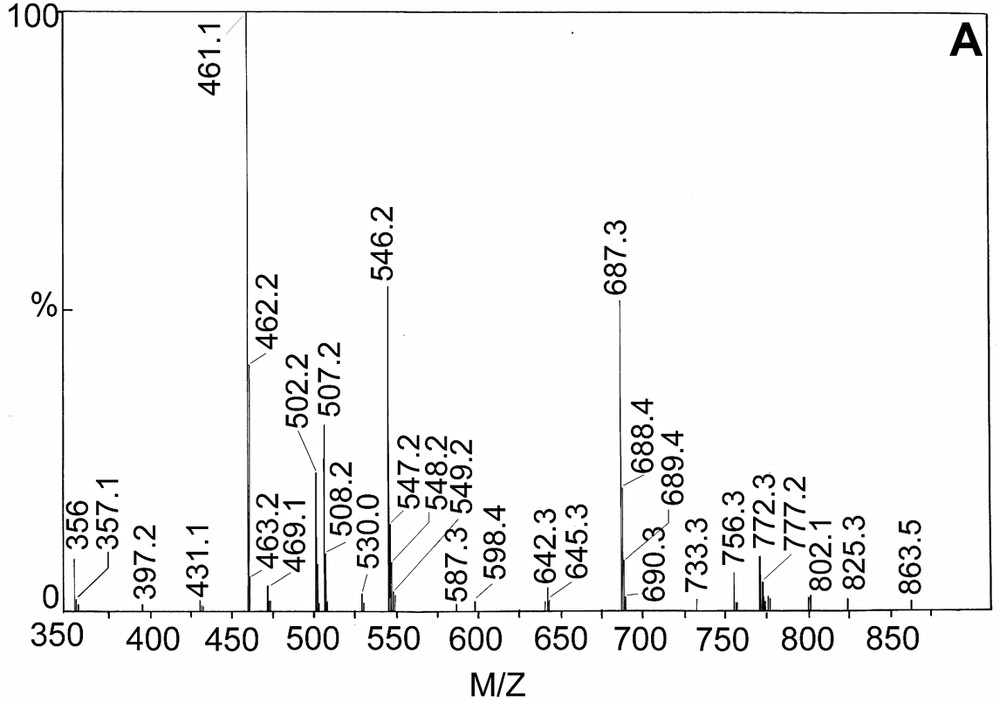
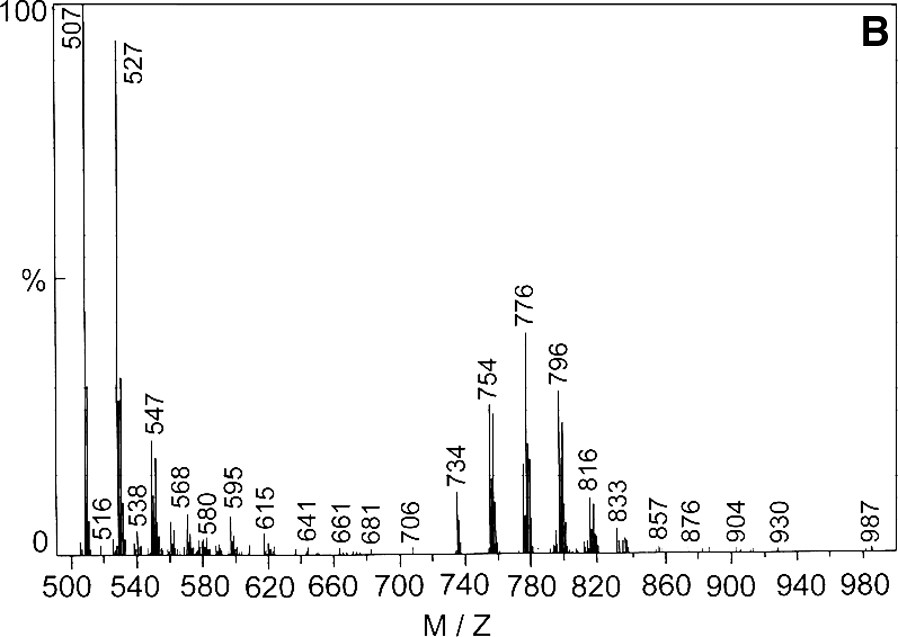
(A) Electrospray mass spectra of complex [MnIII(BOTHA)3] and (B) [MnIII(BOCPHA)3].
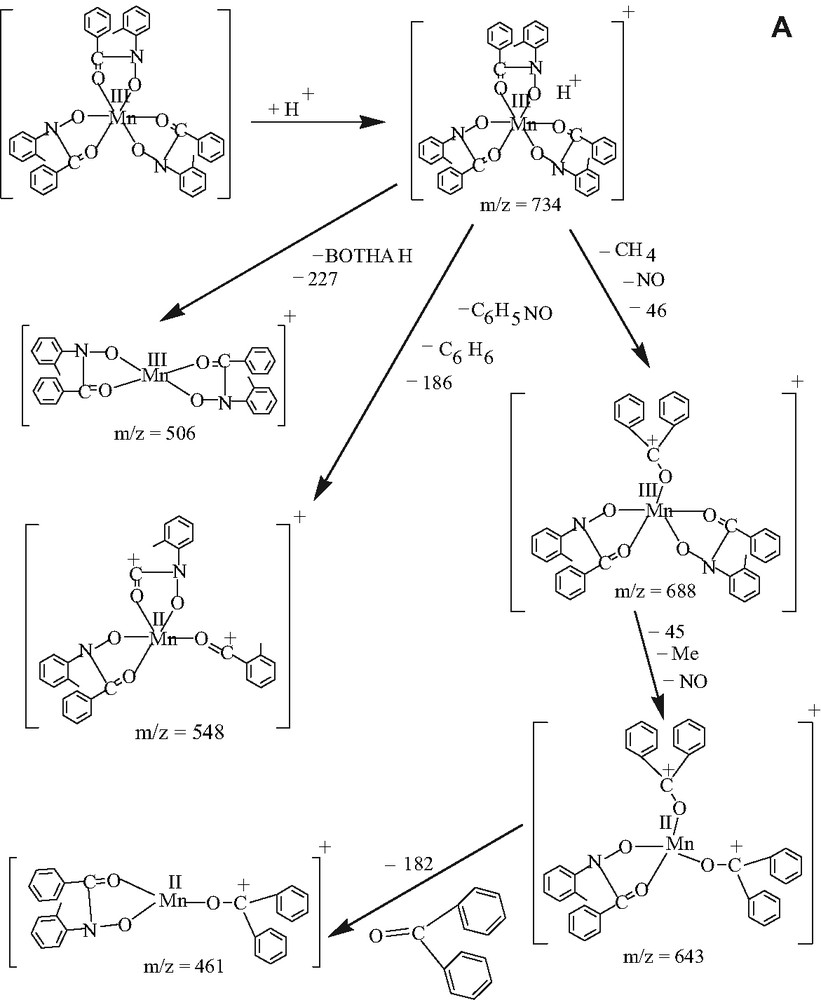
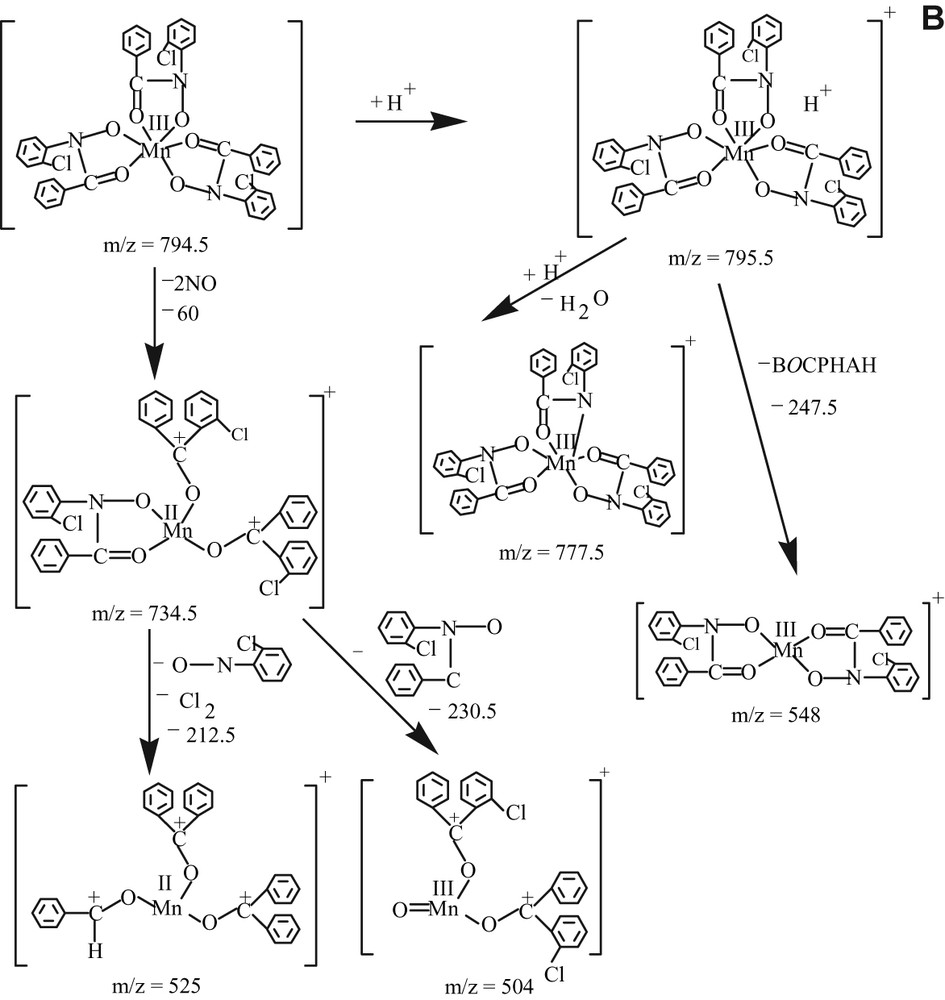
(A) The fragmentation pattern of electrospray mass spectrum of complexes [MnIII(BOTHA)3] and (B) [MnIII(BOCPHA)3] in the positive mode.
4.6 Catalytic epoxidation
The efficient method for epoxidation of olefins [22–25] is extremely important for organic synthesis and in developmental research of interest to the industries. The Mn(III)-hydroxamates 2 and 3 as catalyst, and NaHCO3 buffer solution [26] as co-catalyst is an ideal combination and the efficiency of epoxidation is shown in Table 1 and Table 2. A comparison of yield % (for a particular substrate : catalyst ratio), TON (turnover number = ratio of the mols of product obtained to moles of catalyst used), and TOF (TON h–1) obtained by our method (Tables 1 and 2) with many other Mn(III) catalyzed epoxidation work [22–25,27–29] clearly shows that our method is much more efficient than the said earlier methods. However, we have not yet undertaken a comprehensive work on the kinetics and mechanism of these epoxidation reactions. Earlier workers [30] have demonstrated that a highly active intermediate, viz. MnV = O was responsible for the conversion of to . But in this present case the reaction of HCO3– with H2O2 is known [31] to generate HCO4– (permonocarbonate) which is much more reactive than H2O2 and is expected to attack (immediately) the metal center producing MnV = O or other more potential activated group at a much faster rate and hence TON and TOF become so impressive.
5 Concluding remarks
The fast aerial oxidation of MnII complexes to MnIII only with sterically hindered hydroxamic acid, inhibits the polymerization reaction, which is evidently slower than the oxidation. So, the manganese-hydroxamate systems display a property whereby the relative speed of reactions decides the oxidation state of metal ion. Moreover, these two Mn(III) complexes are obtained purely due to steric effect. This is an interesting case: steric effect slowed down the polymerization reaction whereupon Mn(II) got enough time to be oxidized to Mn(III). Both of these Mn(III)-hydroxamates are quite effective complexes for epoxidation of olefins.
Acknowledgements
Financial assistance from the Council of Scientific and Industrial Research, New Delhi, is gratefully acknowledged. Thanks are also due to CDRI, Lucknow, for doing Electrospray-Mass spectrum and Bose Institute, Calcutta, for EPR spectrum. We also thank DST, Govt. of India for funding Agilent 6890N Gas chromatograph which has been used for the catalytic studies. We also thank Ms. Soma Sen for being involved in this work at the preliminary stage.
Vous devez vous connecter pour continuer.
S'authentifier