

1 Introduction
Endosperm is a seed storage tissue formed within the angiosperm embryo sac from a second fertilization of the central cell. Generally, endosperm cells are triploid, rich in cellular reserves and are compactly arranged without intercellular spaces. Reserves are stored in the form of carbohydrates, protein and lipids, although specific ratios of these components vary depending on the species. The carbohydrate reserve storage in endosperm is composed usually of starch, galactomannans, glucomannans, galactans and arabinogalactans with proportions varying from one species to another [1,2].
In a preceding study, we described the isolation and characterization of arabinan rich polysaccharides from seed endosperm of the fruit of Opuntia ficus-indica (OFI) Cactaceae [3]. The present communication describes the extraction with sodium hydroxide of xylans from this endosperm, their fractionation and characterization.
2 Results and discussion
2.1 Extraction and purification of xylan
The pectic polysaccharides were extracted by hot water and chelating solution from the cell wall material (CWM) of seed endosperm according to our previous work [3]. The residue was extracted sequentially by cold alkaline solution and hot alkaline solution. Two major polysaccharidic fractions were obtained. Cold alkaline soluble fraction (CASF) precipitated by ethanol after neutralization and hot alkaline soluble fraction (HASF) recovered after precipitation occurring during neutralization of hot alkaline extract (Fig. 1). The yield and sugar composition of CWM, CASF and HASF are given in Table 1.
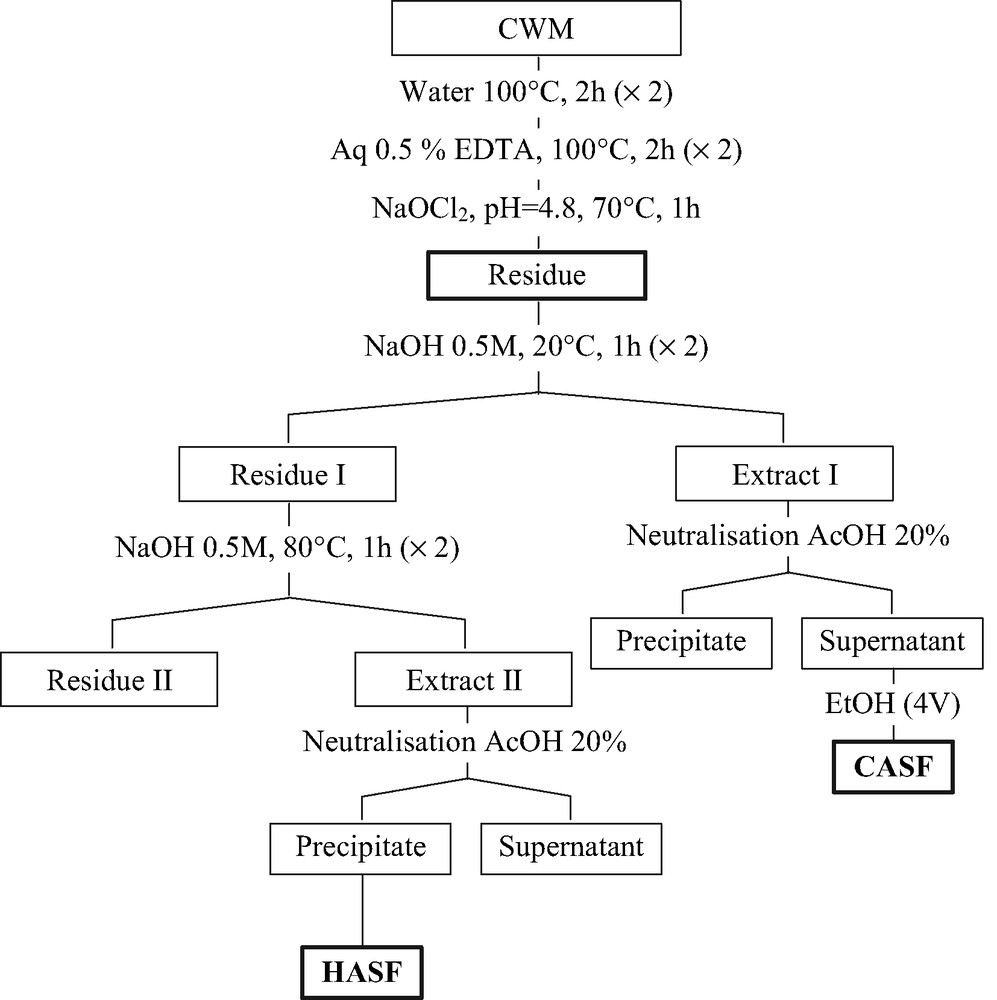
Scheme of isolation of polysaccharides from seed endosperm of OFI.
Yield and sugar composition of CWM, CASF and HASF
| Fraction | Yield a | Uronic acid b | Neutral sugars b | ||||||
| Rha | Glc | Gal | Ara | Xyl | Man | Fuc | |||
| CWM | 84.5 | 35.6 | 3.3 | 29.3 | 8.4 | 12.3 | 4.5 | 1.6 | – |
| CASF | 7 | 26.0 | 3.8 | 2.1 | 4.1 | 37.2 | 3.7 | tr | – |
| HASF | 4.2 | 0 | – | – | – | – | 97 | – | – |
a As % of endosperm dry matter.
b Expressed in relative weight percentages.
The CASF was fractionated by ion exchange chromatography and the buffer eluted fraction (CASF1*) was then purified by size-exclusion chromatography to give a homogenous polysaccharide noted CASF1.
The HASF, which was insoluble in water, was purified by solubilization in 1% NaOH solution and precipitation with Fehling solution to give purified fraction noted HASF1.
2.2 Sugar composition and methylation analysis of xylans
A H2SO4 hydrolyzate of CASF1 revealed that xylose was the major component with a small amount of fucose (xylose and fucose molar ratio of 12:1). The uronic acid content was estimated by a colorimetric method to be 22% (w/w) [4], which corresponded to a molar ratio of xylose to uronic acid of 6:1. The water insoluble fraction HASF1 was composed exclusively of xylose. The sugar composition of CASF1 and HASF1 are reported in Table 2.
Sugar composition of CASF1 and HASF1
| Fraction | Uronic acid a | Neutral sugars a | ||||
| Glc | Gal | Ara | Xyl | Fuc | ||
| CASF1 | 22 | – | – | tr | 72 | 6 |
| HASF1 | 0 | – | – | – | 99 | – |
a Expressed in relative weight percentages.
Based on the hitherto published results [5–7], we know that the xylans of all higher plants possess β-(1 → 4) linked xylopyranose units as the backbone. Depending on their origin, i.e. Gramineae, Gymnosperms or Angiosperms the backbone is usually substituted with α-d-glucuronic acid, 4-O-methyl-α-d-glucuronic acid and some neutral sugar units such as α-l-arabinose, α-d-xylose and α-d-galactose. The unusual occurrence of fucose in polysaccharides from cell walls of higher plants and the absence of previous evidence for this sugar as a constituent of xylan family prompted us to undertake a more detailed examination. While referring to the literature, the only xylan type polysaccharide containing fucose residues was isolated by Aspinall et al. [8] from Hyptis suaveolens.
The results of methylation analysis are reported in Table 3 and indicated that the backbone of all xylan fractions consisted of (1 → 4) linked xylopyranose. In the CASF1, the results showed the presence, in addition to 2,3-di-O-methyl-xylitol, of O-2 and O-3 substituted xylose confirmed by the presence of 3-O-methyl-xylitol and 2-O-methyl-xylitol. The presence of 2,3,4-tri-O-methyl-fucositol in approximately equal amount with the 2-O-methyl-xylitol confirmed that the fucose units are linked to position O-3 of the xylan backbone. Furthermore, one can notice the poor yields of 3-O-methyl-xylitol due to incomplete hydrolysis of the 4-O-Me-GlcpA-(1 → 2)-Xylp linkage.
Partially methylated alditol acetates of CASF1 and HASF1
| Sugar derivatives | Molar ratio a | Mode of linkages |
| CASF1 | ||
| 2,3-Me2-Xyl b | 79 | →4)-β-Xylp-(1→ |
| 2-Me-Xyl | 6.9 | →3,4)-β-Xylp-(1→ |
| 3-Me-Xyl | 5.5 | →2,4)-β-Xylp-(1→ |
| 2,3,4-Me3-Fuc | 7.1 | T-Fucp |
| HASF1 | ||
| 2,3-Me2-Xyl | 99 | →4)-β-Xylp-(1→ |
a Relative mole ratio.
b 2,3-Me2-Xyl = 1,4-di-O-acetyl-2,3-di-O-methyl-xylitol, etc.
The detection of only 2,3-di-O-methyl-xylitol in the case of HASF1 indicated that it consisted exclusively of (1 → 4) linked xylopyranose units.
2.3 NMR characterization of xylan
The structures of CASF1 and HASF1 were also investigated by NMR spectroscopy. The 1H and 13C NMR spectra of CASF1 recorded in D2O are given in Figs. 2 and 3. The 13C NMR spectrum showed five main signals at δ 102.37 (C-1), 73.45 (C-2), 74.45 (C-3), 77.15 (C-4) and 63.74 (C-5) ppm, corresponding to (1 → 4) linked β-d-Xyl residues. Other less intense signals observed at δ 177.16, 98.30, 72.95, 77.44, 83.03, 73.15 and 60.23 ppm, are characteristic, respectively, of C-6, C-1, C-2, C-3, C-4, C-5 and the methoxyl group of a 4-O-methyl-α-d-glucuronic acid residue. Signals at δ 101.95, 77.41, 72.12, 75.95, 62.39 ppm are characteristics of C-1, C-2, C-3, C-4, C-5, respectively, of β-d-Xyl units substituted with 4-O-methyl-α-d-GlcA in O-2.
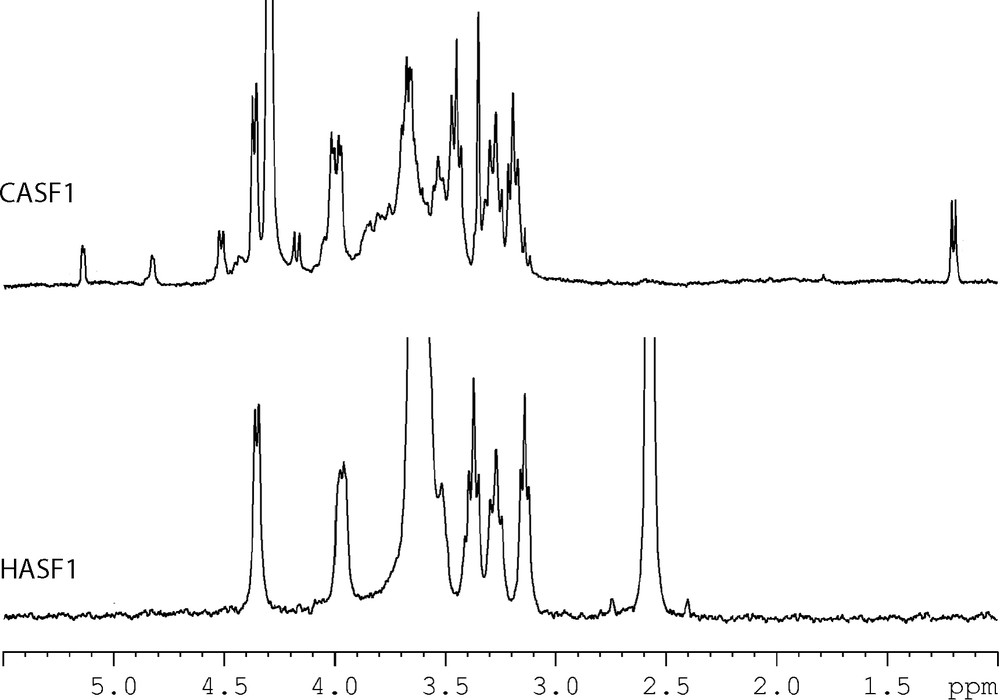
1H NMR spectra of CSAF1 (D2O) and HASF1 (DMSO-d6) at 333 K.

13C NMR spectra of CSAF1 (D2O) and HASF1 (DMSO-d6) at 333 K.
The presence of fucose was also confirmed by the presence in 13C NMR spectrum of characteristic signals at 99.67 (C-1), 68.53 (C-2), 76.24 (C-3), 69.01 (C-4), 61.41 (C-5) and 20.85 (C-6) ppm). Also the xylose residues substituted with fucose are characterized by signals at 103.12 (C-1), 72.27 (C-2), 76.88 (C-3), 76.24 (C-4) and 63.50 (C-5) ppm.
The average integration of all signals for the different sugar residues, in 13C quantitative spectrum, revealed a molar ratio of xylose, fucose to 4-O-methyl-α-d-glucuronic acid, respectively, of 12:1:2.
Examination of the proton spectrum (Fig. 2) of CASF1 showed the relative simplicity of the structure as exhibited by: (i) major signals at δ 4.36 (H-1), 3.97 (H-5eq), 3.60 (H-4), 3.40 (H-3), 3.23 (H-5ax) and 3.14 ppm (H-2), corresponding to non-substituted β-d-Xyl residues; (ii) minor signals at δ 5.14 (H-1), 4.16 (H-5), 3.58 (H-3), 3.46 (H-2), 3.35 (OCH3), 3.12 ppm (H-4), corresponding to 4-O-methyl-α-d-GlcA acid residues, and at δ 4.51 (H-1), 4.01 (H-5eq), 3.60 (H-4), 3.53 (H-3), 3.38 (H-2) and 3.22 ppm (H-5ax) assigned to β-d-Xyl units substituted with 4-O-methyl-α-d-GlcA. The presence of fucose units was also confirmed by the presence of a doublet at 4.83 ppm assigned to H-1 with a small coupling J1,2 = 1.5 Hz characteristic of fucosyl units α-linked and another doublet at 1.19 ppm corresponding to the methyl group.
The proton spectrum showed five doublets in the anomeric region at 4.36 [→ 4)-β-d-Xylp-(1→], 4.51 [→ 1,2)-β-d-Xylp-(→ 4], 4.45 [→ 1,3)-β-d-Xylp-(→ 4], 4.83 [α-Fucp] and 5.14 ppm [4-O-Me-α-d-GlcpA] with an intensity ratio of 9:2:1:2:1, respectively. The proton NMR data confirmed the presence of 12 xylose units per two uronic acid and one fucose residues.
The NMR data for CASF1 are reported in Table 4 and are in good agreement with the structures of (4-O-methyl-α-d-glucurono)-β-d-xylans already described in a number of plants [9–12].
Chemical shift data (333 K) for related residues of CASF1 and HASF1 fractions
| Glycosyl residues | Assignment | |||||
| 1 | 2 | 3 | 4 | 5 | ||
| CASF1 a | ||||||
| →4)-β-d-Xylp-(1→ | 1H | 4.36 | 3.14 | 3.40 | 3.60 | Heq: 3.97 /Hax: 3.23 |
| 13C | 102.37 | 73.45 | 74.45 | 77.15 | 63.74 | |
| →2,4)-β-d-Xylp-(1→ | 1H | 4.51 | 3.38 | 3.53 | 3.60 | Heq: 4.01 /Hax: 3.22 |
| 13C | 101.95 | 77.41 | 72.12 | 75.95 | 62.39 | |
| →3,4)-β-d-Xylp-(1→ | 1H | 4.45 | na | na | na | na |
| 13C | 103.12 | 72.27 | 76.88 | 76.24 | 63.50 | |
| α-l-Fucp-(→ | 1H | 4.83 | 3.65 | 3.80 | 3.70 | na/CH3: 1.19 |
| 13C | 99.67 | 68.53 | 76.24 | 69.01 | 61.41/CH3: 20.85 | |
| 4-O-Me-α-d-GlcpA-(→ | 1H | 5.14 | 3.46 | 3.58 | 3.12 | 4.16 /OCH3: 3.35 |
| 13C | 98.30 | 72.95 | 77.44 | 83.03 | 73.15 | |
| OCH3: 60.23 /C-6: 177.16 | ||||||
| HASF1 b | ||||||
| →4)-β-d-Xylp-(1→ | 1H | 4.26 | 3.05 | 3.28 | 3.53 | Heq: 3.88 /Hax: 3.18 |
| 13C | 102.34 | 73.32 | 74.59 | 76.02 | 63.81 |
a In ppm relative to the signal of internal acetone in deuterium oxide, at 2.1 ppm (1H) or at 31.5 ppm (13C).
b In ppm relative to the signal of internal acetone in DMSO-d6, at 2.1 ppm (1H) or at 31.5 ppm (13C).
The 1H and 13C NMR spectrum of HASF1 recorded in DMSO-d6 are given in Figs. 2 and 3. The 13C NMR spectrum showed only five signals at δ 102.34 (C-1), 73.32 (C-2), 74.59 (C-3), 76.02 (C-4) and 63.81 (C-5) ppm, corresponding to (1 → 4) linked β-d-Xyl residues. Also the 1H NMR spectrum showed the simplicity of the structure as exhibited by only six signals at δ 4.26 (H-1), 3.05 (H-2), 3.28 (H-3), 3.53 (H-4), 3.18 (H-5ax) and 3.88 (H-5eq). The NMR data reported in Table 4 for HASF1 are in good agreement with the structure of linear (1 → 4)-β-d-xylan already described in guar seed husk [13].
3 Experimental
3.1 Materials
Fresh mature prickly pear fruits of OFI were collected in November 2000 from the experimental station plantation located in the vicinity of Marrakech (Morocco). The harvested fruits were washed, carefully hand-peeled and the pulp containing the seeds was mixed for a few minutes in a mixer grinder. The seeds were recovered from the resulting pulp juice by straining through a metallic strainer and cleaned by several washings in distilled water. After drying, they were cracked in an analytical grinder for a few minutes and the endosperm was recovered as a fine powder after sieving on a 60 mesh sieve.
3.2 Analytical methods
Uronic acid content was determined according to Blumenkrantz and Asboe-Hansen's [4] method. Neutral sugars were analyzed, after H2SO4 hydrolysis, by GLC as their corresponding alditol acetates, using a Packard and Becker 417 instrument coupled to a Hewlett–Packard 3380 A integrator. Glass columns (3 mm × 2 m) packed with 3% SP 2340 on Chromosorb W-AW DMCS (100–120 mesh), or 3% OV 17 on the same support were used. The carboxyl groups of the d-galactosyluronic acid were reduced according to the method of Taylor and Conrad [14]. The carboxyl-reduced and the neutrals samples were methylated twice by the Hakomori procedure, as described by Jansson et al. [15]. The partially methylated carbohydrates were then converted into their alditol acetates by successive treatments with NaBH4 and pyridine-Ac2O and analyzed on a fused-silica widebore column (30 m × 0.53 m) half bonded with SP-2380. Peak identification was based on retention times using partially methylated alditol acetates standard and confirmed by GLC by using a SP 2380 capillary column (0.32 mm) coupled to a Nermag R1010C mass spectrometer. Peak areas were corrected by using the molar response factors according to Sweet et al. [16].
3.3 Preparation of CWM
Fats, waxes and oils (11 wt.% of dry material) were removed from endosperm powder by refluxing in a Soxhlet apparatus during 24 h with 38:62 toluene/EtOH. The CWM was prepared by enzymatic digestion according to Habibi et al. (unpublished results).
3.4 Isolation of xylan
CWM was sequentially treated by water (2 × 2 h at 100 °C) and aqueous solution of calcium chelator agent 0.5% EDTA (2 × 2 h at 100 °C) and finally sodium chlorite treatment was performed to remove residual protein and lignin. The bleached residue was extracted by cold alkaline solution (0.5 M, 2 × 1 h at 20 °C) and hot alkaline solution (0.5 M, 2 × 1 h at 80 °C). All extracts were neutralized (pH ≈ 5–6) and the precipitates were recovered by centrifugation. The supernatant was precipitated with EtOH (4 vol). The extraction scheme is given in Fig. 1.
3.5 Ion exchange chromatography
A sample of CASF (450 mg) was suspended in 100 ml of 0.05 M phosphate buffer (pH 6.3) and loaded onto a column of DEAE–Trisacryl M (phosphate form), which was eluted sequentially with phosphate buffer and then with a NaCl gradient (0.125–1 M) in the same buffer. The buffer eluted fraction was then dialyzed against distilled water and freeze-dried to give a polysaccharide fraction CASF1*.
3.6 Purification of xylan
The buffer eluted fraction CASF1* was purified by size-exclusion chromatography on a Shodex-OHpak B-803 (7.5 × 500 mm) column, eluted at 1 ml/min flow rate with 0.05 M NaNO3 solution, and at room temperature. The column effluent was monitored using a refractive index detector. The salts were removed by dialysis and the solution freeze-dried, to give the purified fraction CASF1.
The insoluble fraction HASF was purified by solubilization in 1% NaOH solution and precipitation with Fehling solution according to Jones and Stoodley [17].
3.7 NMR spectroscopy
1H experiments were recorded on a Bruker Avance 400 spectrometer (operating frequency of 400.13 MHz). 13C NMR experiments were obtained on the same spectrometer (operating frequency: 100.57 MHz). CASF1 was examined as solution in D2O at 333°K in 5 mm o.d. tube (internal acetone 1H (CH3) at 2.1 ppm and 13C (CH3) at 31.5 ppm relative to Me4Si). HASF1 was examined recorded as solution in DMSO-d6 at 333°K in 5 mm o.d. tube (internal acetone 1H (CH3) at 2.1 ppm and 13C (CH3) at 31.5 ppm relative to Me4Si).
Acknowledgements
We acknowledge the financial help of the ‘Comité mixte franco-marocain’ (‘Action intégrée’ 236/SVS/00).