

1 Introduction
Eleutherobin [1] and sarcodyctyins [2,3] (1 and 2, Fig. 1) are natural diterpenic compounds extracted from corals and sponges. Eleutherobin (1) exhibits antitumor properties and its mechanism of action was found similar to that of paclitaxel (Taxol®) [4].

Only a few total syntheses of eleutherobin [5,6], sarcodyctyins [7] and analogs [8–10] have been described up to now. We have recently got involved into a project of convergent synthesis of the title compounds: our retrosynthetic approach, which involves the functionalization of suitably substituted 5-methylfuran-2(5H)-ones, followed by an intramolecular Diels–Alder cyclization as the key step, is outlined in Scheme 1.
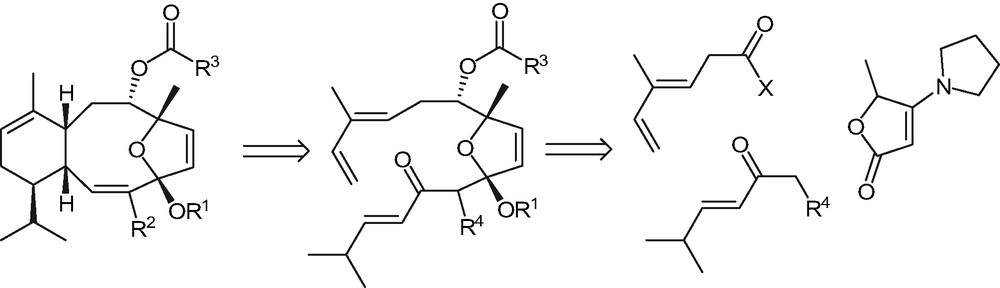
We have recently shown [11] that acylation of 5-methyl-4-(pyrrolydin-1′-yl)-furan-2(5H)-one, followed by the reduction of the intermediate, afforded the corresponding syn products in good diastereoselectivity and yield, thus making the introduction of the necessary diene moiety possible [12]. We hereby report on the main results of a preliminary investigation aimed at introducing the dienophile residue by addition reactions of suitable enolates to the required lactone. This study was carried out on the racemic model systems 3a and 3b (cf. Schemes 2 and 3).
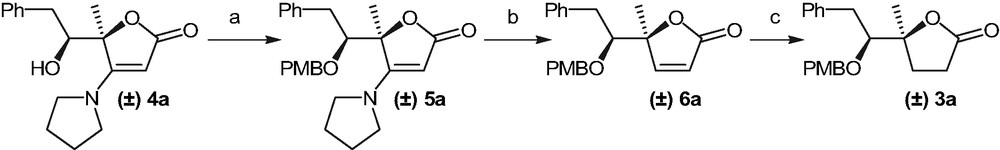
Reagents and conditions: (a) KOH, PMBBr, CH2Cl2, TBAI, 90%; (b) (i) NaBH3CN, AcOH; (ii) mCPBA, CH2Cl2, aq. NaHCO3, 98% (two steps); (c) NaBH4, CoCl2·6 H2O, EtOH quant.
2 Results and discussion
The starting materials 3a and 3b used in the present study were prepared in four steps from syn disubsituted furanones 4 [11]. Protection of the hydroxyl group of 4a as a p-methoxybenzyl ether gave 5a, which was transformed into furanone 6a upon a known reduction-elimination procedure. Reduction of 6a afforded 3a in quantitative yield.
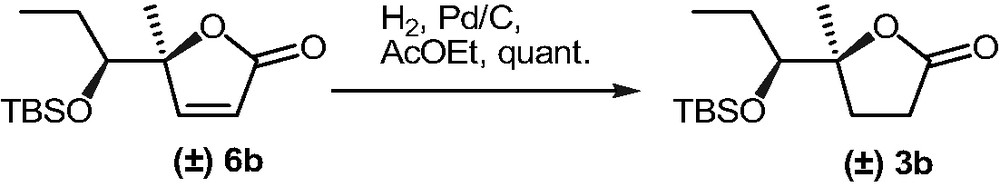
Similarly, 3b was obtained by hydrogenation of known 6b [11] (Scheme 3).
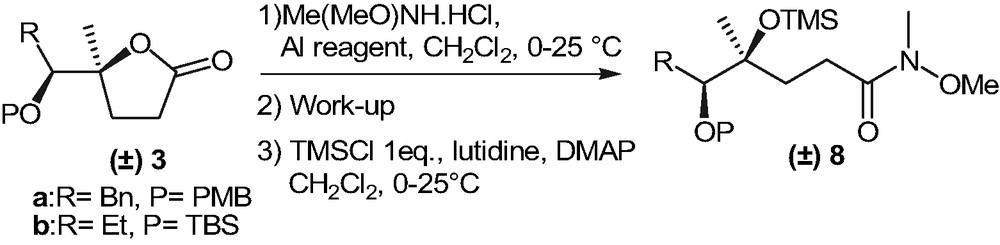
Attempts to react 3a and 3b with lithium enolate of (E)-5-methylhex-3-en-2-one failed, probably because of the acidity of protons at the α position to the lactone carbonyl group [13]. Consequently we considered activating 3a and 3b towards nucleophilic addition through the conversion of the lactones into the corresponding Weinreb amides 7 (Scheme 4).
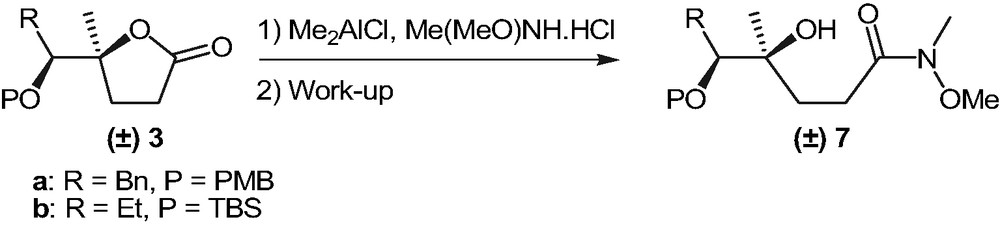
Despite the followed procedure (Me(MeO)NH·HCl in the presence of Me2AlCl) was described as particularly useful for the preparation of Weinreb amides of 4,4-disubstituted lactones [14], all attempts to purify 7 afforded the desired products in low yield together with the starting materials 3. The latter appeared to be formed by recyclization of 7 since TLC analysis of the reaction mixtures prior to work-up always showed the complete disappearance of the precursors 3. Further efforts were thus made in order to convert 7 into the corresponding trimethylsilyl derivatives 8 (Scheme 5). The main collected data regarding the preparation of compounds 8 are summarized in Table 1.
Preparation and protection of Weinreb amides of lactones 3
| Entry | Lactone | Al reagent | Work-up procedure | 8/3 ratioa | 8% yieldb,c |
| 1 | 3b | Me2AlCl | A | 50/50 | 36 (b) |
| 2d | 3b | Me2AlCl | A | 30/70 | 25 (b) |
| 3 | 3b | Me2AlCl | B | 70/30 | 40 (b) |
| 4 | 3b | Me2AlCl | C | 99/1 | 64 (b) |
| 5e | 3a | Me3Al | C | 60/40 | 50 (a) |
| 6f | 3a | Me3Al | C | 80/20 | 58 (a) |
a 8/3 molar ratio was evaluated by integration of selected 1H NMR resonance signals of crude material.
b isolated yield, starting from 100 mg of 3, after purification of crude material by flash chromatography.
c MS, 1H and 13C NMR analyses were in agreement with the structures.
d two equivalents of TMSCl were used.
e THF was used as solvent.
f a CH2Cl2 solution of Me3Al was used.
The conversion of 3b into 8b was strongly affected by the work-up procedure prior to the treatment with the silylating agent. Basic hydrolysis (work-up procedures A and B) of the reaction mixtures (Table 1, entries 1–3) afforded 8b in low isolated yields (25–40%) and, in each case, the formation of slightly variable amounts of starting material 3b was observed. Further efforts to carry out the hydrolysis under weak acidic conditions afforded similar results. Nevertheless when the crude reaction mixture was treated with silica gel, filtered and engaged into the following protection step (work-up procedure C) the cyclization of the intermediate amide 7b into 3b was minimized and a satisfactory 64% yield of 8b was obtained (Table 1, entry 4).
It has to be underlined that the separation of 8b and 3b was always possible, thus allowing the recovery of the starting material. For the synthesis of 8a, Me2AlCl caused the elimination of the p-methoxybenzyl group of 3a and had to be replaced by Me3Al. The more sluggish reaction, followed by the same treatment successfully used for the preparation of 8b, finally afforded 8a in acceptable yield (Table 1, entry 6).
The protected Weinreb amides were then tested for their reactivity towards various nucleophiles. Reaction between 8 and lithium enolate of (E)-5-methylhex-3-en-2-one failed, even when three molar equivalents of the nucleophile were used. Another synthetic sequence, involving an addition reaction of the ketophosphonate 9 followed by a Horner–Emmons condensation with iso-butyraldehyde was attempted, according to the retrosynthesis described in Scheme 6.
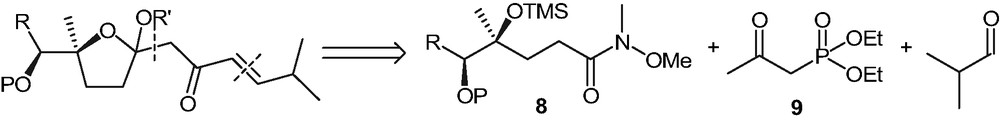
The reaction between 8a and the lithium dienolate of diethyl 2-oxopropylphosphonate (Scheme 7) caused the rapid disappearance (TLC) of the starting material. After usual work-up, a mixture of inseparable products 10–12 was obtained, whose structures (Scheme 7) were assigned on the basis of mass spectrometry analyses. It seems reasonable to ascribe the formation of 11 to the instability of TMS protecting group under the strongly basic conditions necessary for the dienolate formation and the formation of 12 to the cyclization of 11, followed by water elimination.
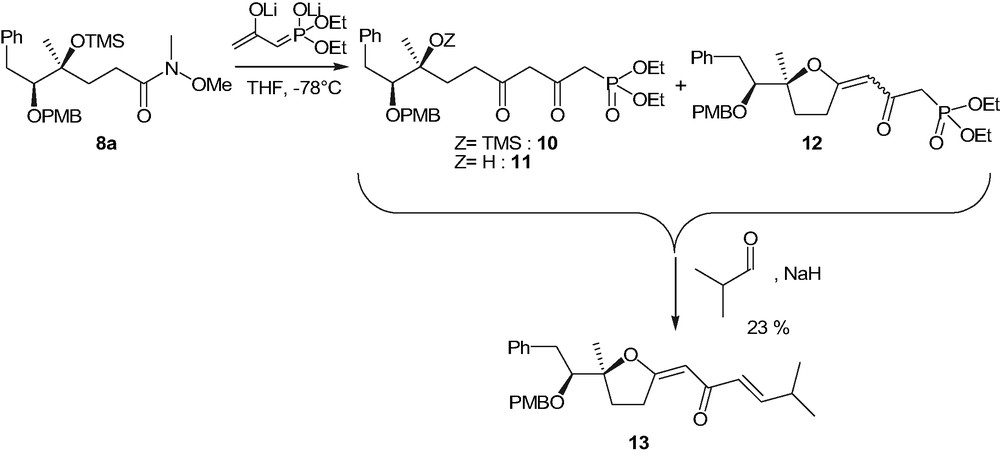
Anyway, the treatment of the mixture of 10–12 with iso-butyraldehyde, under Horner–Emmons reaction conditions, afforded (1E,3E)-1-{5-[1-(4-methoxybenzyloxy)-2-phenylethyl]-5-methyldihydrofuran-2-ylidene}-5-methyl-hex-3-en-2-one 13 (Scheme 7) as a unique isolated product in 23% yield over the two steps.
The formation of intermediates 11 and 12, along with product 13, clearly enlightened the necessity of replacing TMS in 8 by a more stable and selectively removable protecting group. Studies are in progress to identify new suitable reaction conditions for the preparation of such a compound and to functionalize the enol ether group of 13.
Acknowledgements
We are grateful to the CNRS for financing SS with a post-doctoral fellowship.