

1 Introduction
Molecular electronics is an actively emerging field of research in chemistry and in physics. The wealth of open questions related to electrical conductivity through single molecules, collective effects and the functionalities of these molecules has opened a wide area of frontier research [1–4].
Most of the molecules investigated so far for the application as molecular wires are phenylethynyl oligomers, which are rigid and have more or less planar structures [5,6]. As a consequence some problems may arise:
- • the rigid, two dimensional, and highly aromatic structure is prone to aggregation of two or more molecular wires by some kind of π,π stacking. Therefore, it is unclear, whether acquired electronic data result from just one single molecule or from a less defined aggregate;
- • for an exact fit of a molecular wire between two electrodes the electrode distance and the length of the molecular wire must be adjusted.
Our approach makes use of the fact that ferrocene can be regarded as a three dimensional analogue of benzene and will therefore be less prone to aggregation. Due to the unhindered rotation around the cyclopentadienyl–iron–cyclopentadienyl axis, it is also expected that the structure would behave like a foldable ruler, which can adjust its effective length as required by the distance between the electrodes. Recently Dinglasan et al. [7] reported the self organization of 1,1′-ferrocene dicarboxylic acid in as tunnel junctions between aluminum and aluminum oxide layers. The exploitation of the conformational flexibility of ferrocene derivatives in the context off molecular recognition has been pointed out by Constable [8] as early as 1991.
Here we describe the synthesis of 1,1′-bis[(4-thioacetylphenyl)ethynyl]ferrocene (3), the first example of the class of molecules we are interested in.
2 Synthesis
The monosubstituted phenylethynyl ferrocene with a thioacetyl functionality was synthesized in 1995 via a Sonogashira coupling by Hsung et al. [9]. The synthesis of 3 is based on convergent methodologies [10–12]. The deprotection of 1 to 1,1′-diethynylferrocene confirmed its reported instability owing to spontaneous polymerization even under exclusion of water and oxygen [13]. Our synthetic approach to 3 is illustrated in Scheme 1.
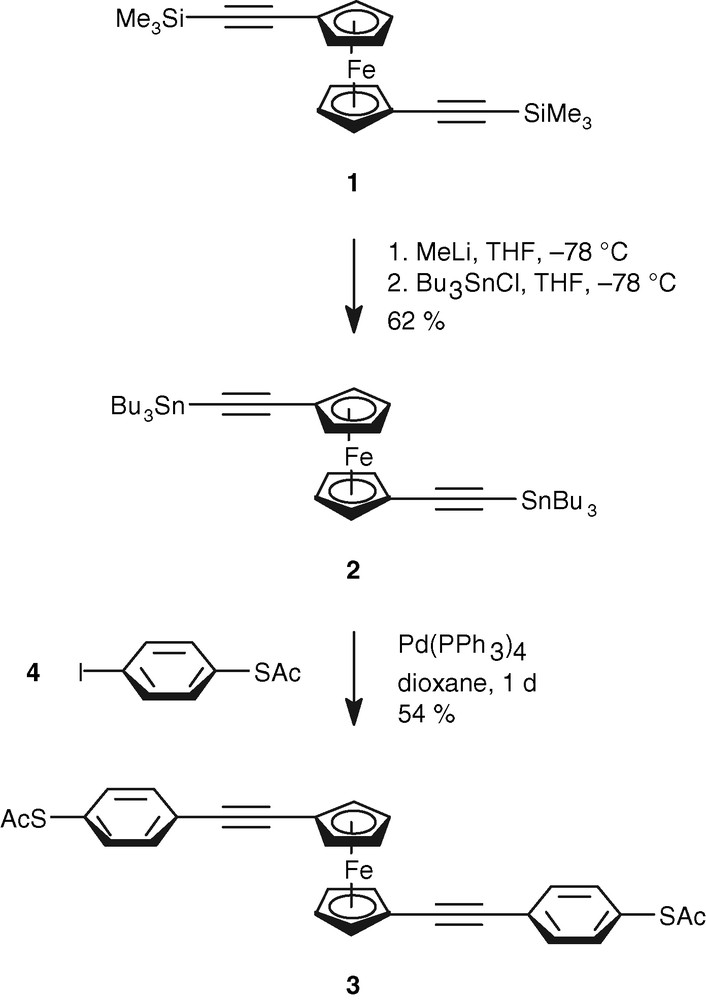
Synthesis of 1,1′-bis[(4-thioacetylphenyl)ethynyl]ferrocene (3).
1,1′-Bis(tributylstannylethynyl)ferrocene (2) was synthesized from the trimethylsilyl-protected diethynylferrocene 1 [10] under N2 by lithiation with 2.1 equiv. of methyl lithium followed by addition of Bu3SnCl in THF at –78 °C. The purification is problematic, because it is not easy to separate the excess of tributylstannyl chloride. In contrast to the synthesis of the corresponding trimethylstannyl derivative [13] the less volatile tributylstannyl chloride had to be removed either by vacuum distillation (150 °C, 12 mbar) or by column chromatography. As the presence of a small amount of tributylstannyl chloride does not affect the envisaged Stille coupling, we decided for practical reasons to remove residual stannyl halide after the coupling reaction. The coupling with the alkynyl component is known to proceed 100 times faster than that with an alkyl group [14].
The target compound 3 was synthesized under usual Stille conditions with 2.2 equiv. of 1-iodo-4-thioacetylbenzene (4) [15] catalyzed by 2.5 mol% of tetrakis(triphenylphosphane)palladium(0) in refluxing dioxane for 20 h in moderate yields (54%) (Scheme 1). Residual tributylstannyl chloride was removed by evaporation of the mixture in vacuo to dryness followed by treatment of the crude product with acetonitrile, from which the stannyl halide was extracted with hexane. As 3 is only sparingly soluble in hexane, after three extractions the material isolated was sufficiently pure as to allow further purification by column chromatography on silica gel [petroleum ether → petroleum ether/dichloromethane (2:1)]. 3 was obtained in 54% yield.
3 is an air- and moisture-stable orange–red solid, which melts at 140 °C. As so far no crystals suitable for an X-ray structure analysis could be obtained, 3 was characterized spectroscopically. The IR-spectrum shows the typical stretching frequencies for the alkynyl (2207 cm–1) and carbonyl functional groups (1704 cm–1), and the high resolution mass spectrometric data are in full accord with 3. In the 1H NMR spectrum one AA′BB′ line system at δ = 4.0–4.5 is diagnostic for the symmetry around the ferrocene nucleus. The 13C NMR data also confirm the constitution of 3 with the resonances of the acetylenic carbon atoms appearing at δ = 87.2 and 89.4. Semiempiric calculations indicate the anti conformation of 3 to be the most stable one [14–16]. Cyclic voltammetry (CV) data of 3 are summarized in Table 1. 3 is reversibly oxidized at 0.377 V vs. Fc/Fc+ at 200 mV s–1 and at 0.410 V vs. Fc/Fc+ at 500 mV s–1. The potential is more anodic than that of ferrocene thus indicating that the alkynyl substituents act as electron acceptors towards the ferrocene moiety. The variation of E1/2 with the scan rate indicates a slow electron transfer, which might bee due to the structural dynamics of 3. The E1/2 values obtained are in full accord with those of other acceptor substituted ferrocenes such as 1,1′-diacetylferrocene (0.49 V vs. Fc/Fc+) or ferrocenylcarbonitril (0.31 V vs. Fc/Fc+) [16]. For Fc/Fc+ ΔE was determined to be 0.322 V at a scan rate of 200 mV s–1 and 0.309 V at 500 mV s–1. The comparatively small change for ferrrocene indicates that the changes observed for 3 are intrinsic to the compound.
CV data of 3. 0.2 mol l–1 Bu4N+ PF6– in MeCN, c = 0.0005 mol l–1, T = 296 K, reference electrode: silver wire ø 1 mm, potentials vs. Fc/Fc+
| v [mV/s] | Epc [V] | Epa [V] | ΔE [V] | E1/2 [V] |
| 500 | 0.327 | 0.494 | 0.167 | 0.410 |
| 200 | 0.338 | 0.415 | 0.077 | 0.377 |
3 Discussion and prospects
A convenient method for the preparation of a functionalized dialkynylferrocene 3 is outlined in this paper. A rotation around the Cp–Fe–Cp axis will allow the molecule to adopt a variety of conformations with different distances between the protected thiol groups. Having a use as a molecular wire in mind, this allows the molecule to adjust its effective length according to a given electrode distance. In this context one might expect a U shaped conformation with either one of the thiol groups being bound to the same surface. Although this possibility cannot be excluded yet, one has to take into account that the U shaped conformation is not calculated to be the most stable one [17–19]. The report of Dinglasan et al. [7] does not indicate such a U shaped conformation in the case of 1,1′-ferrocene dicarboxylic acid. In addition, the problem is less likely to arise with higher homologues like 6 for steric reasons.
3 can be regarded as the first representative of a class of compounds in which more than one ferrocene fragment are connected by alkynyl and phenylene units. By this way linear rigid sections of a molecule are connected by ferrocene type hinges resulting in a structure comparable to a foldable ruler. These structures could even better adjust their effective length to the needs of a given geometry of electrodes. We plan to build up such structures by making use of a building block principle put forward by Tour [1]. For example, we envisage the coupling of 2 with 4-bromo(trimethylsilylethynyl)benzene followed by stannylation with MeLi/Bu3SnCl to give 5 (Scheme 2). Coupling with 1,1′-diiodoferrocene followed by attachment of a trimethylsilylethynyl substituent at the other cyclopentadienyl rings followed by a reaction sequence analogous to that in Scheme 1 will finally lead to the foldable ruler molecule 6 (Scheme 2).
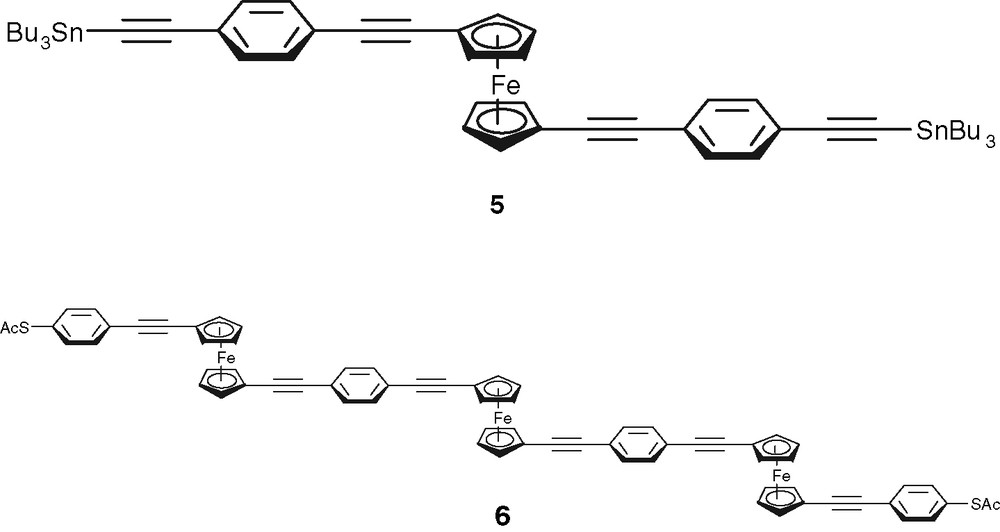
Higher homologues of 2 and 3 to be accessed by the building block principle.
3 and its higher homologues will be anchored at gold surfaces in order to investigate their electronic transport properties by AFM and STM measurements.
4 Experimental
All preparations were carried out under an atmosphere of nitrogen using standard Schlenk techniques. THF was freshly distilled from Na/K alloy/benzophenone under nitrogen prior to use.
4.1 1,1′-Bis(tributylstannylethynyl)ferrocene (2)
At –78 °C 1.6 g (4.2 mmol) of 1 in 160 ml of THF was slowly added to a solution of 8 ml of methyllithium (1.5 M in diethyl ether, 12 mmol). After complete addition the mixture was stirred at 22 °C for 16 h, after which a –78 °C cold solution of 5.6 ml (6.8 g, 21 mmol) of tributylstannyl chloride in 40 ml of THF was added. After stirring at 22 °C for 16 h, the mixture was heated at reflux for 3 h, followed by evaporation of the solvents in vacuo. The crude material was distilled under vacuum to remove excess tributylstannyl chloride. 3.2 g (3.93 mmol, 94%, purity 60%) of 2 was obtained as a dark brown oil. IR (neat): ν(C≡C) 2136 cm–1; 1H NMR (CDCl3, 100.6 MHz): δ 4.17 (t, 4H, Cp), 4.40 (t, 4H, Cp); 13C NMR (C6D6, 50.0 MHz) δ 11.3 (SnCH2), 14.0 (CH3), 27.6 (SnCH2CH2), 29.4 (SnCH2CH2CH2), 68.7 (C–CCp), 71.7 (HCCp). 73.7 (HCCp), 89.6 (≡Csn), 108.5 (CC≡); MS: m/z 812 (M+).
4.2 1,1′-Bis[(4-thioacetylphenyl)ethynyl]ferrocene (3)
A mixture of 900 mg (1.1 mmol) of 2, 69 mg (0.06 mmol, 2.5 mol%) Pd(PPh3)4 and 670 mg (2.4 mmol) of 1-iodo-4-thioacetylbenzene (4) in 25 ml of dioxane was stirred at 100 °C for 24 h, followed by evaporation of the solvents in vacuo. The crude material was treated with 10 ml of acetonitrile, washed three times with 10 ml of hexane each, and was purified by deactivated silca-gel column chromatography (petroleum ether, petrol ether → dichloromethane 1:2) to yield 328 mg (0.6 mmol, 54%) of 3 as an orange–red solid (m.p. 140 °C). Data for 3: IR (neat): ν(C≡C) 2206 cm–1, ν(C=O) 1695 cm–1; 1H NMR (C6D6, 200.1 MHz): δ 1.84 (s, 6H, CH3); 4.00 (t, 4H, Cp), 4.51 (t, 4H, Cp), 7.12 (d, 4H, C6H4), 7.31 (d, 4H, C6H4); 13C NMR (C6D6, 50.0 MHz): δ 29.7 (CH3), 67.9 (C–CCp), 71.1 (H–CCp), 73.4 (H–CCp), 87.2 (≡C), 89.4 (≡C), 125.3 (C–CAr), 127.1 (C–CAr), 132.1 (H–CAr), 134.6 (H–CAr), 191.5 (C=O); HRMS: m/z 534.0412 (calcd. 534.0410).
Acknowledgements
We thank Dr. F. Janetzko and cand. chem. R. Haunschild for the theoretical calculations. We are indebted to the University of Hannover and the Volkswagen Foundation for financial support.