

1 Introduction
Octahedral clusters of transition elements are well known to build [(M6Li8)La6] and [(M6Li12)La6] units [1–3] with halogens or/and chalcogens depending whether the inner ligands are located on bridging or capping position (i = inner and a = apical according to Schäfer and Schnering notation [1]). The metal–metal bonding states of the molecular orbital diagrams are fulfilled by 24 and 16 valence electrons per cluster for face-capped [(M6Li8)La6] and edge-bridged [(M6Li12)La6] units, respectively (VEC = 24 and VEC = 16). The octahedral cluster chemistry of molybdenum is dominated by the [(Mo6Li8)La6] unit except for oxides owing to the small size of oxygen ligand compared to those of other chalcogens and halogens. Indeed, more generally the octahedral cluster oxides of transition elements are always built up from [(M6Oi12)Oa6] units [4–7, M = Mo, Nb, Ta, and W].
In the present work, we report the synthesis and the crystal structure of Cs3Mo6Br13O that constitutes the first molybdenum octahedral cluster oxyhalide. Astonishingly, contrarily to previously reported M6 oxyhalides and oxides, the oxygen ligand is found to be in a μ3-face-capping position. It turns out that the crystal structure, determined by single crystal X-ray diffraction, is built up from discrete face-capped [Mo6Bri6Li2Bra6]3– anionic units (L = 0.5 Br + 0.5 O) in which two inner positions -located on the threefold axis of the unit- are randomly occupied by one bromine and one oxygen whereas the other ligand positions are fully occupied by bromine. The caesium cations randomly occupy two close crystallographic positions generated by the A–B–C–A′–B′–C′ close-packed stacking of the units. The structural findings will be discussed by comparison with previously reported Nb6, Ta6 and W6 oxyhalides [8,9], Cs2Mo6Br14 [10], Rb3Re6S7Br7 [11] as well as Mo3O13 based oxides [12].
2 Experimental part
2.1 Synthesis of Cs3Mo6Br13O
Few single crystals of Cs3Mo6Br13O were obtained as byproduct in a reaction designed to synthesize Cs2Mo6Br14 [10]. A stoichiometric mixture (0.5 g) of CsBr (Alfa Aesar 99.9%) and MoBr2 prepared according to procedure described in [10], was ground, formed as a pellet and placed into a silica tube (o.d. 9 mm, i.d. 7 mm, length 70 mm). Once sealed under vacuum, the tube was heated at 950 °C for 3 days. Few red colored single crystals of Cs3Mo6Br13O suitable for X-ray diffraction studies were directly obtained during the synthesis, while the major bulk product was Cs2Mo6Br14. EDS analysis, using a scanning electron microscope JEOL JSM 6400 equipped with a microprobe EDS OXFORD LINK ISIS, evidenced that the crystals contained the expected elements in a ratio close to the stoichiometry of the Cs3Mo6Br13O formula (atom % Cs/Mo/Br: experimental: 14.7/26.5/58.7; theoretical: 13.6/27.3/59.1). Oxygen element has been clearly detected but not quantified owing to the limited accuracy of the measurement in the presence of heavy elements. Several attempts in order to obtain bulk compound, using in particular MoO3 as starting material, have failed. Indeed, the possibility to use MoO3 as oxygen source has been invalidated. The presence of oxygen in the single crystals of the title compound may be explained by some oxygen contamination during the preparation of MoBr2. New experiments are now in progress in order to obtain Cs3Mo6Br13O in higher yield.
2.2 X-ray single crystal diffraction studies
Single crystal X-ray diffraction data of Cs3Mo6Br13O were collected at room temperature on a Nonius KappaCCD X-ray area-detector diffractometer using Mo Kα radiation (λ = 0.71073 Å) (Centre de diffractométrie de l'université Rennes-1, France). Details of data collection and structure refinement are reported in Table 1. The data processing was performed by the Kappa CCD analysis software [13] and a spherical absorption correction was applied [14]. The calculation of E-statistics [15] performed on the data suggested a centrosymmetric space group. Among the possible space groups deduced from the observed systematic extinctions, the refinement procedure has allowed to retain unambiguously the R-3c centrosymmetric space group. Direct methods (program SIR97 [16]) yielded a first partial structural solution, including the molybdenum cluster, inner ligands and cesium cations. After several cycles of refinements (SHELXL-97 program [17]), it turned out that the bromine inner ligand (Br1) located on the 12c Wyckoff positions did not fully occupy its position contrary to the two other bromine atoms located on two 36f positions. Furthermore, an electronic density peak remained close to Br1 at a distance from molybdenum corresponding to a Mo–O bondlength. Oxygen O1 was then introduced on this residue with the same atomic displacement parameters as Br1. The sum of the occupancies of Br1 and O1 was restricted to the value corresponding to a fully occupied 12c position. Afterwards, the first two restraints were progressively relaxed during the convergence, leading to final positions in agreement with reliable Mo–(Br, O) interatomic distances. However, at this stage, Cs1 exhibited unrealistic anisotropic atomic displacement parameters and a residual electronic density peak remained close to this position. Cs2 was then positioned on this peak with the same isotropic atomic displacement parameter as Cs1 and the sum of the occupancies of Cs1 and Cs2 was restricted to the value corresponding to a full 18e Wyckoff position. Subsequently these restraints were progressively relaxed during the convergence, leading to final reliable anisotropical displacement parameters for Cs1 and Cs2. The final refinements led to the following developed formula Cs2.94(6)Mo6Bri6(Bri0.98(2)Oi1.02(2))Bra6 that corresponds to the Cs3Mo6Bri7OiBra6 formula within the s.u.'s. The defined Cs3Mo6Bri7OiBra6 stoichiometric formula is corroborated by electronic consideration since it corresponds to a valence electron count per cluster of 24 (versus a fractional value of 23.92 for Cs2.94(6)Mo6Bri6(Bri0.98(2)Oi1.02(2))Bra6). The atomic positional parameters and the isotropic equivalent atomic displacement parameters are reported in Table 2. Relevant interatomic distances for discussion of this structure are reported in Table 3.
Crystal and refinement data for the structure determination of Cs3Mo6Br13O
| Empirical formula | Cs2.94(6)Mo6Br12.98(2)O1.02(2) |
| Formula Weight | 2019.8 |
| Space group | Rc No. 167 |
| a (Å) | 15.5784(2) |
| c (Å) | 19.5103(5) |
| V (Å3) | 4100.5(1) |
| Z | 6 |
| Dcalc (g cm–3) | 4.91 |
| Crystal dimensions (mm3) | 0.137 × 0.114 × 0.110 |
| Total reflections collected | 26429 |
| Unique reflections | 2854 |
| Rint (all reflections) | 0.0791 |
| μ (mm–1) | 25.602 |
| T (°C) | 20 |
| λ (Å) | 0.71069 |
| Observed reflections [I > 2 σ (I)] | 2854 |
| Refined parameters | 48 |
| R1,a [I > 2 σ (I)] | 0.0458 |
| wR2,a all data | 0.1148 |
| Δρmin/Δρmax (e Å–3) | –1.875/2.346 |
a R1 = Σhkl∣Fo–Fc∣/Σhkl∣Fo∣; wR2 = [Σhkl[w(Fo2–Fc2)2]/Σhkl[w(Fo2)2]]1/2.
Positional and displacement parameters (Å2) for Cs3Mo6Br13O
| Atom | Position | x | y | z | Occupancy | Ueq |
| Mo1 | 36f | 0.90592(3) | 0.90307(3) | 0.05615(2) | 1 | 0.01613(9) |
| Br1 | 12c | 0 | 0 | 0.16562(12) | 0.492(7) | 0.0189(5) |
| O1 | 12c | 0 | 0 | 0.1339(11) | 0.508(7) | 0.019(2) |
| Br2 | 36f | 0.80270(4) | 0.99380(4) | 0.05307(3) | 1 | 0.02527(13) |
| Br3 | 36f | 0.78351(5) | 0.77780(5) | 0.14676(3) | 1 | 0.03648(16) |
| Cs1 | 18e | 0.7947(3) | 0 | 1/4 | 0.663(16) | 0.0565(7) |
| Cs2 | 18e | 0.8235(8) | 0 | 1/4 | 0.317(16) | 0.0375(15) |
Interatomic distances (Å) in Cs3Mo6Br13O
| Mo6Br13O unit | |
| Mo1–Mo1 | 2.5778(6) × 2 |
| 2.6488(7) × 2 | |
| Mo1–O1 | 2.125(15) × 1 |
| Mo1–Br1 | 2.603(2) × 1 |
| Mo1–Br2 | 2.6191(6) × 1 |
| 2.6245(6) × 1 | |
| 2.6282(6) × 1 | |
| Mo1–Br3 | 2.6170(7) × 1 |
| Cesium environment | |
| Cs1–O1 | 3.93(1) × 2 |
| Cs1–Br1 | 3.597(4) × 2 |
| Cs1–Br2 | 3.8468(6) × 2 |
| Cs1–Br3 | 3.457(4) × 2 |
| 3.822(2) × 2 | |
| 3.932(2) × 2 | |
| Cs2–O1 | 3.562(16) × 2 |
| Cs2–Br1 | 3.204(11) × 2 |
| Cs2–Br2 | 3.853(1) × 2 |
| Cs2–Br3 | 3.679(3) × 2 |
| 3.777(4) × 2 | |
| 3.831(11) × 2 | |
| Shortest interatomic distances between units | |
| O1–O1 | 4.53(3) |
| O1–Br1 | 3.91(2) |
| Br1–Br1 | 3.293(3) |
3 Results and discussion
The Cs3Mo6Br13O oxybromide crystallizes in the trigonal system (space group R-3c) and is isostructural with Rb3Re6S7Br7 [11]. The structure is built up from discrete [(Mo6Bri6Li2)Bra6]3– anionic units (L = 0.5 O + 0.5 Br) of –3 symmetry (Fig. 1). The molybdenum cluster is face-capped by eight inner ligands in such a way that two inner ligand positions located on the ternary axis (12c Wyckoff position) are randomly occupied by bromine (Br1) and oxygen (O1) atoms, whereas the six other inner ligand positions and the six apical ligand positions (36f Wyckoff positions) are fully occupied by bromine (Br2 and Br3, respectively). The value of the refined Br1:O1 ratio is equal to 0.49(1):0.51(1) and corresponds, within the s.u.'s, to a 0.5/0.5 random occupancy of the 12c position by Br1 and O1. It results in a molybdenum cluster in which two opposite triangular faces, perpendicular to the ternary axis, are randomly capped by oxygen and bromine whilst the six other triangular faces, around the ternary axis, are capped by bromine. Notice that the structure of Cs3Mo6Br13O is closely related to that of Rb3Re6S7Br7. However, in Rb3Re6(Si5Bri)Si2Bra6, six inner ligand positions are randomly occupied by sulfur and bromine whilst the two inner ligand positions, lying on the ternary axis, are fully occupied by sulfur. Moreover, the rubidium cation in Rb3Re6(Si5Bri)Si2Bra6 fully occupies its cristallographic positions. The Mo1–Mo1 bond lengths from the Mo3 triangular faces randomly capped by Br1 and O1 are smaller than the Mo1–Mo1 bond lengths from the Mo3 triangular faces fully capped by bromine (2.5778(6) Å and 2.6488(7) Å, respectively). This finding must be attributed to the discrepancy between the ionic radii of bromine and oxygen ligands [18]. The Mo1–O1 bond lengths (2.125(15) Å) and Mo1–Mo1 bond lengths from the Mo3 triangular faces randomly capped by Br1 and O1 (2.5778(6) Å) are close to the corresponding bond lengths reported in molybdenum oxides based on Mo3O13 units containing μ3-Oi as for instance Zn2Mo3O8 or Zn3Mo3O8 [12 and reference therein]. Let us point out that in the latter series the number of valence electrons per cluster, that strongly influences the metal–metal bond length (2.524(2) Å and 2.580(2) Å, respectively), has been found to be for 6 and 8, respectively. The Mo1–Bri1, Mo1–Bri2 and Mo–Bra3 distances vary within the range 2.603(2) Å–2.6282(6) Å and are only slightly larger than those observed in Cs2Mo6Br14 (average Mo–Bri = 2.601 Å). Indeed, the single noticeable evolution when going from a [Mo6Br14]2– to a [Mo6Br13O]3– unit is a decrease of the average Mo–Mo bond length (2.6348(7) Å to 2.6133(7) Å) in relation with the presence of a μ3-Oi face-capping oxygen. This is the reverse situation to what has been observed in [Nb6L18] units going from [Nb6Cl18]4– to [Nb6Cl17O]5– in KLuNb6Cl18 and Cs2LuNb6Cl17O, respectively [8 d]. In this case the presence of one disordered oxygen among the twelve inner ligands of the [Nb6L18] unit does not have any steric influence on the metal–metal bondlentghs. On the other hand, the Nb–Cl interatomic distances are larger in the oxychloride than in the chloride in particular the Nb–Cla one. For higher oxygen content in the unit, an ordering occurs between oxygen and chlorine ligands. This leads to shorter Nb–Nb oxygen-bridged bondlength than Nb–Nb chlorine-bridged ones and to a modification of the electronic properties. Recall that for [Nb6L18] based oxyhalides, when the number of inner oxygen ligands increases, the Nb–Oi antibonding contribution at the HOMO level (a2u symmetry) increases and becomes preponderant for three oxygens per unit leading to an overall nonbonding character for this a2u level [19]. Consequently, the usual VEC for the (Nb6Cl12 – xOx) cluster core is found to be 16 or 15 for x < 3 whereas this value is found to be 14 for x ≥ 3.
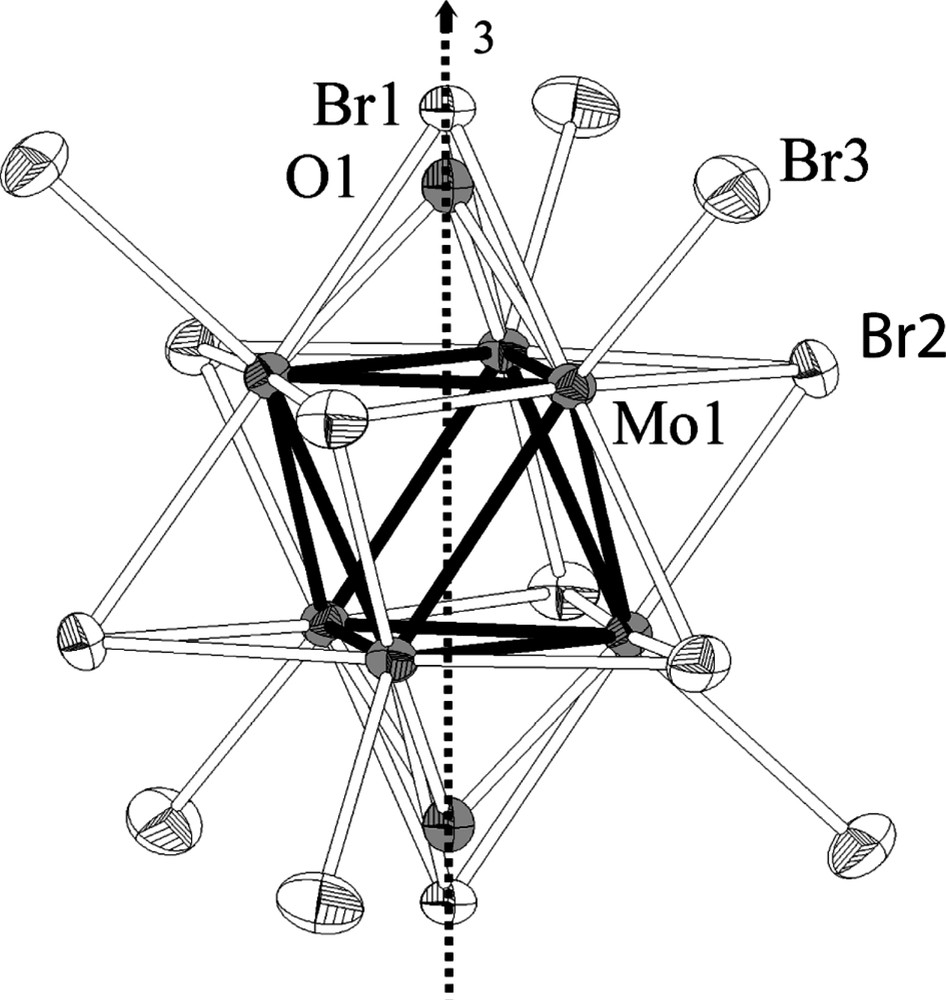
The [(Mo6Bri6Li2)Bra6]3– unit with labelled atoms. Only one of the two O1 and Br1 positions is locally occupied. Displacement ellipsoids are shown at the 50% probability level.
The [(Mo6Bri6Li2)Bra6]3– units are arranged according to a A–B–C–A′–B′–C′ stacking (Fig. 2). The layers are strongly interpenetrated resulting in short distances between the L1 inner ligands located on the ternary axis that belong to adjacent units of layers A and A′. The O1–Br1 interatomic distance is 3.91(2) Å, whereas the Br1–Br1 one of 3.293(3) Å is shorter than the sum of the Br– ionic radii (dBr––Br– = 3.92 Å). This feature implies an alternated distribution of Br1 and O1 ligands along the threefold axis of the structure. The Cs1 and Cs2 caesium cations randomly occupy two different 18e Wyckoff positions with the following refined occupancy: 0.66(2) and 0.32(2), respectively. They are separated by 0.449(13) Å and are surrounded by four cluster units (Fig. 3), belonging to four different layers of the stacking (A, B, C, A′) that build a distorted tetrahedron. The Cs1 10 coordination site (Fig. 3) is built up from two apical ligands and six inner ligands belonging to two cluster units from layers A and A′ and from two additional apical ligands belonging to two cluster units from layer B and C. It results in six Cs1–Bra interatomic distances that vary within the range 3.457(4) Å–3.932(2) Å, two Cs1–Bri interatomic distances of 3.8468(6) Å and two Cs1–Bri/Oi interatomic distances of 3.597(4) Å and 3.93(1) Å, respectively. Cs1 and Cs2 being surrounded by the same four cluster units, their coordination site is almost the same with, however, different interatomic distances. Moreover, the short Cs2–Br1 interatomic distance of 3.204(11) Å (lower than the sum of the ionic radii of Cs+ and Br–) clearly indicates a correlation between the occupancy of Cs2 and those of O1 and Br1. In other words, it means that the occupation of the Cs2 site implies an empty Br1 site and an occupied O1 site. It results in six Cs2–Bra interatomic distances that vary within the range 3.679(3) Å–3.831(11) Å, two Cs2–Bri interatomic distances of 3.853(1) Å and two Cs2–Oi interatomic distances of 3.562(16) Å.
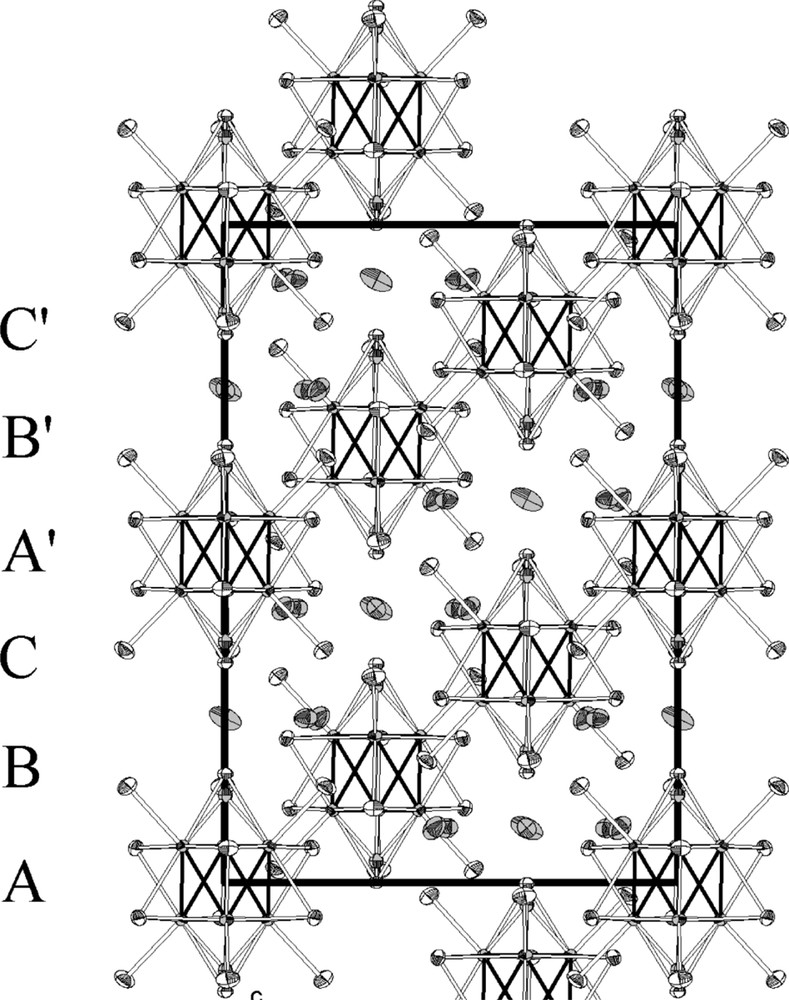
Projection of the structure of Cs3Mo6Br13O along the [010] direction. Displacement ellipsoids are shown at the 50% probability level.
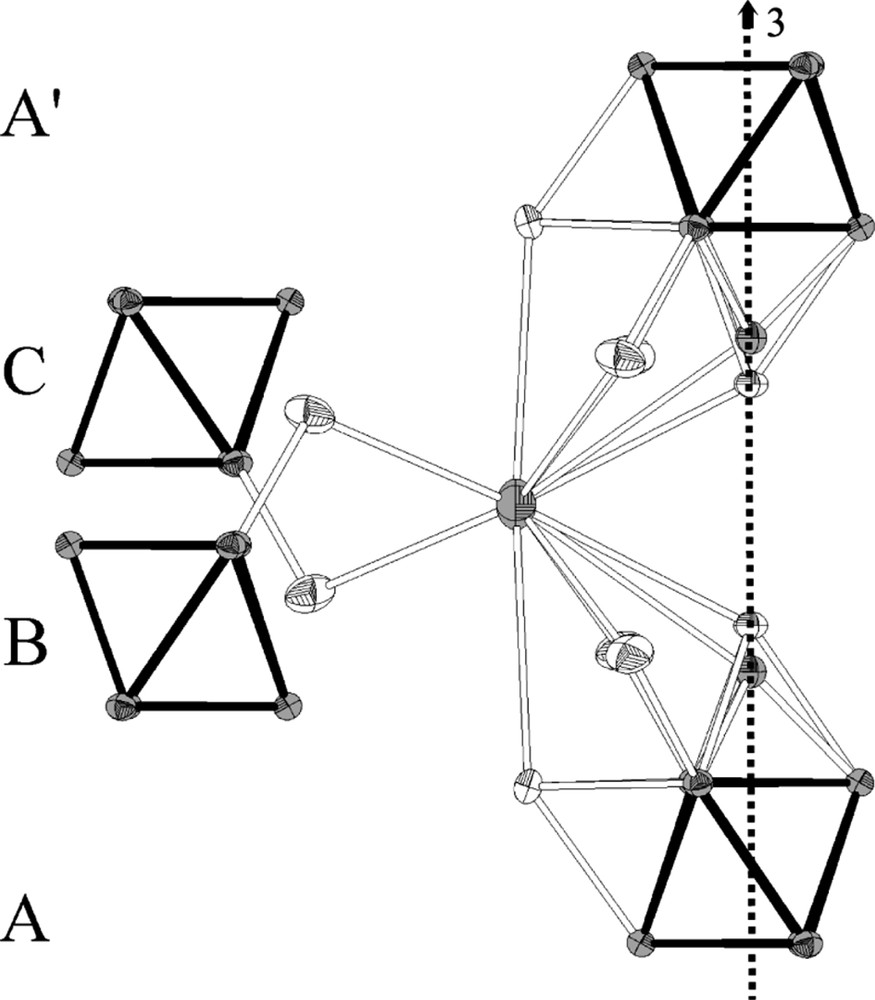
Cs1 environment. The threefold axis is represented as a dotted arrow. Displacement ellipsoids are shown at the 50% probability level.
4 Concluding remarks
The title compound synthesized by solid-state route exhibits for the first time a molybdenum octahedral cluster with a μ3-face-capping oxygen. Beyond the increase of the anionic charge on the cluster unit, the main finding going from [Mo6Br14]2– to a [Mo6Br13O]3– unit is a decrease of the average Mo–Mo bondlength (2.6348–2.6133). The increase of the O/Br ratio on the inner positions of the [M6L14] unit should affect significantly their structural and electronic properties but it could also lead to [Mo6L18] unit based compounds. Let us recall that in the case of tungsten, despite a W6 cluster chemistry also dominated by [W6Li8La6] units, some compounds based on [W6Li12La6] units have been reported. This concerns the W6Cl18 [20] and the ternary AxW6Cl18 [21] chlorides as well as three oxychlorides [9], based on edge-bridged α- or β- [W6O6Cl12]2– isomers and [W6O7Cl11]3– units.
5 Supplementary materials
Further details of the crystal structure investigation can be obtained from the Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (fax: (49) 7247-808-666; e-mail: crysdata@fiz.karlsruhe.de) on quoting the depository numbers CSD 414368.
Acknowledgements
The ‘Centre de diffractométrie de l'université Rennes-1’ is acknowledged for the data collection on the Nonius KappaCCD X-ray diffractometer. In particular, the useful advice of Dr. T. Roisnel (LCSIM, Rennes) is gratefully acknowledged. The ‘Centre de microscopie électronique a balayage de l'université Rennes-1’ is thanked for EDS analyses. We are indebted to the French Research Ministry for financial support (ACI Nanosciences 2001– No. 18-01 contract) as well as ‘Fondation Langlois’.