

1 Introduction
Technetium complexes containing N-donor ligands are widely utilized as diagnostic radiopharmaceuticals, in e.g. commercially known Ceretec (99mTcVO-d,l-HMPAO), Neurolite (99mTcVO-LL-ECD), Technescan (99mTcVO-MAG3) and Technescan Q12 (99mTcIII-furifosmin), all containing Schiff base and related amine type ligands [1–3]. Similarly, radio-rhenium, the 5d analog of technetium, finds application in therapeutic radiopharmacy [4]. However, non-radioactive rhenium is also utilized to imitate technetium chemistry on a macro scale and has been pursued in this way for the past two decades or more. Thus, structural effects induced by amine type ligands in rhenium chemistry, as well as the co-ordination mode of ethylenediamine and functionalized derivatives thereof, are important facets in the ongoing investigation of the co-ordination chemistry of model radiopharmaceuticals.
We have previously investigated different aspects of the trans dioxo complexes, with the general structure of trans-[MO2(L4)]n–, M = Mo(IV), W(IV), Tc(V), Re(V), Os(VI), and related systems, evaluating structural and reactivity correlations for a range of ligands L. It was shown that a 12 order-of-magnitude in reactivity in these systems exists, ranging from the very rapid proton exchange, to the slower hydroxo and aqua substitution and the extremely slow equatorial ligand substitution [5,6].
As an extension of the above-mentioned study, the functionalization of the ethylenediamine type ligands was investigated, and the current study specifically involved the substitution of one of the hydrogen atoms on one amine nitrogen by a pendant ethyl group. This manipulation was investigated to determine what effects–if any–would be induced on the co-ordination mode of the diamine as well as to investigate the chirality introduced by the stereochemical amine centers. Possible additional structures were identified and optimized by density-functional (DFT) methods and correlated with the solid-state data in these classic 18-electron, d2 dioxo complexes.
2 Experimental section
2.1 Materials
All experiments were carried out in air and doubly distilled water was used in all the preparations unless otherwise mentioned. Potassium perrhenate (KReO4) was purchased from Strem Chemicals and the Eten ligand (N-ethyl ethylenediamine) from the Sigma-Aldrich Chemical Company. All these reagents were used as received without further purification.
2.2 Preparation of complexes
2.2.1 (Bun4N)ReO4
KReO4 (0.5 g, 1.73 mmol) and (Bun4N)Cl (0.625 g, 2.25 mmol) (tetrabutyl ammonium chloride) were separately dissolved in the minimum amount of warm water (ca. 5–10 ml, 70 °C) and the latter was slowly added to the ReO4– solution with stirring. The solution was then cooled in an ice bath and the white precipitate formed was vacuum filtered and dried overnight in a vacuum desiccator over P2O5. Quantitative yield. IR: ν(Re=O) = 909 cm–1.
2.2.2 trans-[ReOCl3(PPh3)2]
trans-[ReOCl3(PPh3)2] was synthesized by a slight variation on the method of Johnson et al. [7]. Triphenylphosphine (5.4 g, 20.6 mmol) was dissolved in glacial acetic acid (80 ml). (Bun4N)ReO4 (0.6 g, 1.22 mmol) was dissolved in concentrated hydrochloric acid (6 ml) and added to the triphenylphosphine solution over a period of ca. 45 min with stirring. Once the addition of the (Bun4N)ReO4 was complete the dropping funnel was rinsed with glacial acetic acid (10 ml) which was added to the reaction mixture. Stirring was continued for 5 h followed by filtration of the light green precipitate. The precipitate was washed with glacial acetic acid (10 ml), diethyl ether (3 × 10 ml) and dried overnight in a vacuum desiccator over P2O5 (yield: 0.91 g, 90%). The IR spectrum of this material was identical to that reported previously [7]. IR: ν(Re=O) = 969 cm–1.
2.2.3 trans-[ReO2(Eten)2]CF3SO3·LiCF3SO3
This complex was synthesized via a slight variation of the procedure reported by Ram and Hupp [8] by using Eten instead of pyridine. trans-[ReOCl3(PPh3)2] (250 mg, 0.3 mmol) was suspended in an acetone (20 ml) and water (0.6 ml) mixture with the Eten (1.058 g, 12.0 mmol), {Re/Eten, 1:~40} weighed directly into acetone and added with stirring to the Re suspension. The mixture was refluxed for 90 min and allowed to cool, and left at room temperature overnight–yielding a crystalline product. Good quality crystals were obtained by dissolving trans-[ReO2(Eten)2]Cl (50 mg, 0.12 mmol) in water (ca. 4 ml) and adding LiCF3SO3 (114 mg, 0.73 mmol). The orange–brown crystals were harvested after 3 days of slow evaporation of the solvent at ambient temperature (yield: 59 mg, 73%). Spectral data: IR: ν(Re=O) = 798 cm–1; 1H NMR: 1.07 ppm (triplet, CH3, 3H, methyl on pendant ethyl) and four multiplets at 2.43, 2.63, 2.86 and 3.15 ppm (3 × CH2, 6H, methylene groups on pendant ethyl, C1 and C2); UV/Vis: λmax, nm (ɛ, M–1 cm–1): 435 (25).
2.3 Spectroscopy
1H NMR spectra were recorded in D2O on a Bruker 300 MHz spectrometer; 1H chemical shifts are reported relative to H2O (4.60 ppm). IR spectra were recorded as KBr pellets on a Hitachi 27050 spectrometer and the UV/Vis spectral changes and absorbance spectra on Hitachi 150-20 and Varian Cary 50 Conc spectrophotometers in a 1.000 ± 0.001 cm quartz cell.
2.4 X-ray crystallography
The reflection data were collected on a 1K SMART Siemens instrument equipped with a CCD area detector, using graphite monochromated Mo–Kα radiation (λ = 0.71073 Å) at ambient temperature (293(2) K). The data were corrected for Lorentz and polarization effects, and the structure was solved with conventional Patterson and Fourier methods and refined by full-matrix least-squares on F2 by using SHELX-97 [9], while the graphic display was done with DIAMOND [10].
All non-hydrogen atoms were refined anisotropically and the positions of the hydrogen atoms were calculated as riding on the adjacent carbon and nitrogen atoms with the bond distances as: methylene C–H as 0.97 Å, methyl C–H = 0.96 Å and amine N–H = 0.89 Å. The densities of the crystals were determined by flotation in iodomethane/benzene.
The general crystal data and refinement parameters for trans-[ReO2(Eten)2]CF3SO3·LiCF3SO3 are presented below (Table 1) while a complete list of atomic co-ordinates, equivalent isotropic parameters, bond distances and angles, anisotropic displacement parameters and hydrogen co-ordinates is given in the Supplementary Material.
Crystal data and refinement parameters for tans-[ReO2(Eten)2]CF3SO3·LiCF3SO3
| Empirical formula | C10H24F6LiN4O8S2Re | μ (mm–1) | 5.451 |
| Formula weight | 699.59 | ρexp (g cm–3) | 1.98 |
| Crystal system | Triclinic | ρcalc (g cm–3) | 1.982 |
| Space group | F(000) | 680 | |
| a (Å) | 8.5978(13) | θ Range (°) | 2.83–26.00 |
| b (Å) | 11.1525(17) | Reflections collected/unique | 6991/4551 |
| c (Å) | 13.2356(20) | R(int) | 0.0246 |
| α (°) | 67.796(2) | Completeness to θ (°; %) | 26.00; 98.9 |
| β (°) | 86.302(3) | Tmax/Tmin | 0.5608/0.1638 |
| γ (°) | 87.009(3) | Data/restraints/parameters | 4551/0°/286 |
| Volume (Å3) | 1172.0(3) | Goodness-of-fit on F2 | 0.963 |
| Crystal size (mm) | 0.52 × 0.30 × 0.12 | Final R indices | R1 = 0.0330 |
| Z | 2 | [I > 2 sigma(I)] | wR2 = 0.0899 |
| Index ranges | –10 ≤ h ≤ 7 | R indices (all data) | R1 = 0.0405 |
| –13 ≤ k ≤ 13 | wR2 = 0.0920 | ||
| –16 ≤ l ≤ 15 | Largest peak/hole (e Å–3) | 1.479/–1.939 |
The supplementary material has been sent to the Cambridge Crystallographic Data Center (CCDC), 12 Union Road, Cambridge CB2 1EZ, UK as supplementary material No. SUP CCDC 270 144 and can be obtained by contacting the CCDC (quoting the article details and the corresponding SUP number).
Results of QM computational procedures will be made available by the authors on request (Table 1).
2.5 Computational details
Calculations were performed using GaussianW 03 [11]. The DFT calculations were carried out using the hybrid functional and basis set: B3LYP [12]/LanL2DZ [13]. The DIAMOND [10] program was used for the data, while the graphs were constructed in MS EXCEL [14]. All isomers presented are local minima on the energy surface, and have been characterized by means of frequency analysis to verify this assumption. The single transition state was identified with a single negative frequency.
3 Results and discussion
Orange–brown crystals of the title compound, suitable for X-ray data collection, were isolated as mentioned previously. trans-[ReO2(Eten)2]CF3SO3·LiCF3SO3 crystallizes with two independent formula units per asymmetric unit in the triclinic space group . Each asymmetric unit thus consists of two independent Re complexes (see Fig. 1), one lithium cation and two CF3SO3– anions. The complex shows a distorted octahedral geometry at the six co-ordinate Re(V) metal center, with the four amine nitrogen atoms in the equatorial plane and the two oxo ligands occupying the axial positions. Both Re atoms are situated on inversion centers, with the Li+ cation and CF3SO3– anions occupying general positions.

Selected interatomic interactions in trans-[ReO2(Eten)2]CF3SO3·LiCF3SO3 (20% probability ellipsoids), showing the crystallographic two independent trans-[ReO2(Eten)2]+ cations. H atoms are set at arbitrary size.
Fig. 1 shows some hydrogen bonding as well as lithium cation interactions in the solid state. The methyl groups on the ethyl chain form hydrogen bonds with the neighboring oxygen and fluoride atoms on the triflate anions and these interactions range from 2.48 to 2.88 Å. The tetrahedral Li+ cations form bridges between the O=Re=O cores of adjacent conformers resulting in a polymeric structure. These Re=O–Li–O=Re interactions are 1.887(8) and 1.940(8) Å (Fig. 1) and compare favorably with the 1.903(7) Å reported for the [TcO2(2,3,2-tet)]+ complex (2,3,2-tet = 1,4,8,11-tetraazaundecane), where similar Li+CF3SO3– interactions were observed [15]. The Li+ cation also has two interactions of 2.006(9) and 1.965(8) Å with two adjacent CF3SO3– anions that are comparable to the average of 1.960(8) Å reported for the above-mentioned [TcO2(2,3,2-tet)]+ complex.
The typical numbering scheme in the trans-[ReO2(Eten)2]+ cation is shown in Fig. 2, with the most important interatomic bond distances and angles reported in Table 2. The difference in the geometries of the two crystallographically characterized trans-[ReO2(Eten)2]+ cations in the title complex is illustrated in Fig. 3 but is discussed in more detail later in the paper.
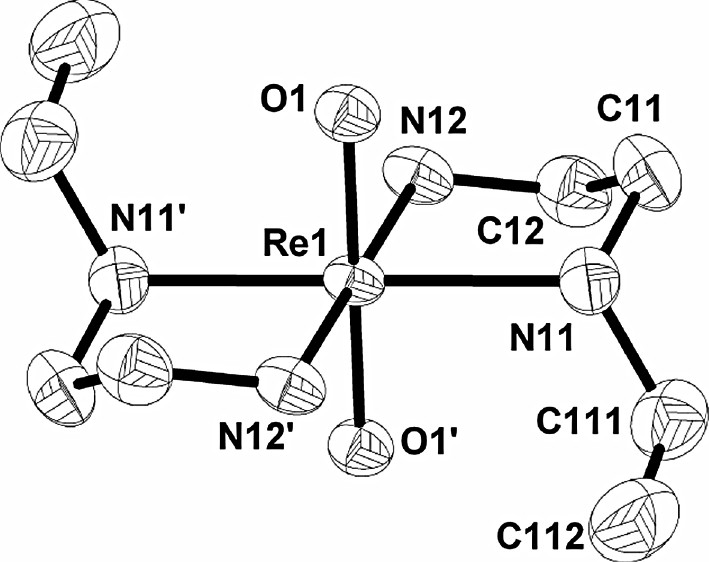
Diamond drawing of the Re(1) cation in trans-[ReO2(Eten)2]CF3SO3·LiCF3SO3 (50% probability ellipsoids), showing the general numbering scheme. The Re(2) cation, anion, Li+ and hydrogen atoms are omitted for clarity. Primed atoms denote those generated by a center of symmetry.
Selected bond lengths (Å) and angles (°) for trans-[ReO2(Eten)2]CF3SO3·LiCF3SO3
| Re(1)–O(1) | 1.764(3) | Re(2)–O(2)#2 | 1.766(3) |
| Re(1)–O(1)#1 | 1.764(3) | Re(2)–O(2) | 1.766(3) |
| Re(1)–N(11) | 2.196(4) | Re(2)–N(21) | 2.186(4) |
| Re(1)–N(11)#1 | 2.196(4) | Re(2)–N(21)#2 | 2.186(4) |
| Re(1)–N(12) | 2.163(4) | Re(2)–N(22)#2 | 2.160(4) |
| Re(1)–N(12)#1 | 2.163(4) | Re(2)–N(22) | 2.160(4) |
| N(11)–C(111) | 1.489(7) | N(21)–C(211) | 1.477(7) |
| N(11)–C(11) | 1.492(6) | N(21)–C(21) | 1.495(8) |
| N(12)–C(12) | 1.467(6) | N(22)–C(22) | 1.511(7) |
| C(11)–C(12) | 1.494(8) | C(21)–C(22) | 1.479(9) |
| C(111)–C(112) | 1.490(9) | C(211)–C(212) | 1.497(9) |
| O(1)–Li | 1.940(8) | O(2)–Li | 1.887(8) |
| Li–O(012) | 1.965(8) | Li–O(021) | 2.006(9) |
| O(1)–Re(1)–N(11) | 89.21(15) | O(2)–Re(2)–N(21) | 90.04(16) |
| O(1)–Re(1)–N(12) | 91.49(14) | O(2)–Re(2)–N(22) | 90.93(16) |
| N(12)–Re(1)–N(11) | 79.21(15) | N(22)–Re(2)–N(21) | 79.69(16) |
| O(1)–Re(1)–O(1)#1 | 180 | O(2)#2–Re(2)–O(2) | 180 |
| O(1)–Re(1)–N(12)#1 | 88.51(14) | O(2)#2–Re(2)–N(22) | 89.07(16) |
| O(1)–Re(1)–N(11)#1 | 90.79(15) | O(2)–Re(2)–N(21)#2 | 89.96(16) |
| N(12)–Re(1)–N(11)#1 | 100.79(15) | N(22)–Re(2)–N(21)#2 | 100.31(16) |
| O(1)#1–Re(1)–N(11) | 90.79(15) | O(2)#2–Re(2)–N(21)#2 | 90.04(16) |
| C(111)–N(11)–Re(1) | 119.5(3) | C(211)–N(21)–Re(2) | 115.5(3) |
| C(11)–N(11)–Re(1) | 107.1(3) | C(21)–N(21)–Re(2) | 109.0(4) |
| C(12)–N(12)–Re(1) | 110.7(3) | C(22)–N(22)–Re(2) | 109.4(3) |
| N(11)–C(11)–C(12) | 109.1(4) | C(22)–C(21)–N(21) | 109.6(5) |
| N(11)–C(111)–C(112) | 112.3(5) | N(21)–C(211)–C(212) | 116.0(5) |
| C(111)–N(11)–C(11) | 111.1(5) | C(211)–N(21)–C(21) | 113.0(5) |
| N(12)–C(12)–C(11) | 107.9(4) | C(21)–C(22)–N(22) | 108.4(5) |
| Re(1)–O(1)–Li | 163.5(3) | Re(2)–O(2)–Li | 158.2(3) |
| O(2)–Li–O(1) | 119.4(4) | O(2)–Li–O(012) | 110.9(4) |
| O(1)–Li–O(012) | 112.2(4) | O(2)–Li–O(021) | 100.3(4) |
| O(1)–Li–O(021) | 105.5(4) | O(012)–Li–O(021) | 106.9(4) |
| Re(1)–N(11)–C(111)–C112) | 65.7(6) | Re(2)–N(21)–C(211)–C(212) | 178.3(5) |
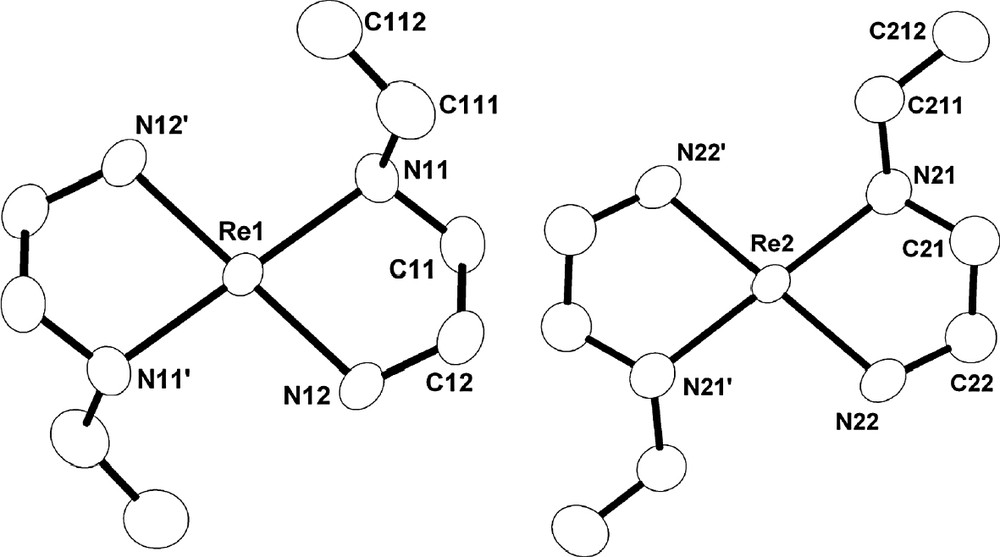
Illustration of the differences between the two independent trans-[ReO2(Eten)2]+ cations, referenced to Re1 and Re2, when viewed along the O=Re=O axis (50% probability ellipsoids). Oxo ligands and hydrogen atoms omitted for clarity.
The average Re=O bond length of 1.765(3) Å is typical of a trans O=Re=O orientation and is well within range of the average for Re=O bonds reported as 1.761(15) Å [16]. The average bite angle of 79.5(2)° is typical for a five-membered chelate and compares well with previously isolated structures. The geometrical parameters observed in the title compound are compared to other similar complexes from literature (see Table 3). Good agreement, with small variations between individual metal centers, exists [17,6,18–21].
Comparative table of average bond lengths, bite angles and IR stretching frequencies for various diamine type dioxo d2 complexes
| Complex | Bond distance (Å) | Ligand bite angle | ν(M=O) | References | ||
| M=O | M–N a | M–Nfunc b | (°) | (cm–1) | ||
| [ReO2(en)2]+ | 1.765(7) | 2.162(9) | – | 80.0(3) | 820 | [17] |
| [ReO2(Eten)2]+ | 1.765(4) | 2.162(4) | 2.191(4) | 79.5(3) | 798 | c |
| [ReO2(DiEten)2]+ | 1.769(2) | 2.172(3) | 2.242(3) | 80.3(1) | 810 | [6] |
| [ReO2(diFcen)2]+ | 1.774(8) | – | 2.204(13) | 79.2(4) | 810 | [18] |
| [TcO2(en)2]+ | 1.747(1) | 2.158(2) | – | 80.37(6) | 833 | [19] |
| [OsO2(en)2]2+ | 1.74(1) | 2.11(1) | – | 80.2(5) | 917 | [20] |
| [OsO(but-en)2]+ | 1.72(2) | 1.99(1) | 1.85(1) d | 79.0(4) | 920 | [21] |
a M–NH2 bond (two protons on amine nitrogen).
b M–Nfunc = functionalized amine bond.
c This work, KBr pellet, average of two complexes.
d Imine bond.
The two functionalized amine nitrogen atoms (containing pendant ethyl groups) are trans with respect to one another, and this phenomenon is currently interpreted to be due to the steric crowding at the metal center. The energy values associated with the different isomers as obtained from DFT calculations are discussed below.
It is evident that the Re–N bonds for the functionalized amine [Re1–N11 = 2.196(4); Re2–N21 2.186(4) Å] are significantly longer than that of the unfunctionalized one [Re1–N12 = 2.163(4); Re2–N22 = 2.160(4) Å]. This average Re–N bond length of 2.162(5) Å for the unfunctionalized N-donor is essentially the same as for the trans-[ReO2(en)2]+ complex (2.162(9) Å [17]) indicating that the lengthening observed for the Re–N21 bond is due to the functionalization incorporated on the mentioned amine nitrogen atom (and the direct effects resulting from this). This lengthening in the Re-functionalized amine bond could be due to the trans influence that these two functionalized moieties exert on each other (thus preferring the trans orientation to cis) as well as to steric crowding.
The Eten ligand has one stereochemical center which results in two such centers for the trans-[ReO2(Eten)2]+ cation. For both Re complexes the N11′/N21 moieties exhibit the R conformation while the N11/N21′ amines both have S conformations. Thus, the individual complexes exhibit RS stereochemistry in the solid state. It is clear that different isomers and conformers can be formed during the synthesis of the title compound.
Similar racemic tendencies in the solid state were observed for related nickel(II) [22–24] and copper(II) [25,26] structures with mono-substituted bidentate amine ligands. In fact, few complexes could thus far be found in the Cambridge Crystallographic Database [27] that show only one conformation (RR or SS) [28]. This suggests a thermodynamic drive toward the RS mode.
An interesting phenomenon in this type of complexes is the orientation of the ethylene backbones relative to each other in the two Eten ligands. By viewing the title compound perpendicular to both the O=Re=O axis and the carbon backbones of the ethylenediamine ligands, the carbon atoms of the ethylene moieties are eclipsed (λδ) and not staggered (δδ + λλ), resulting in a δλ conformation for the chelating amines (Fig. 4) [29].
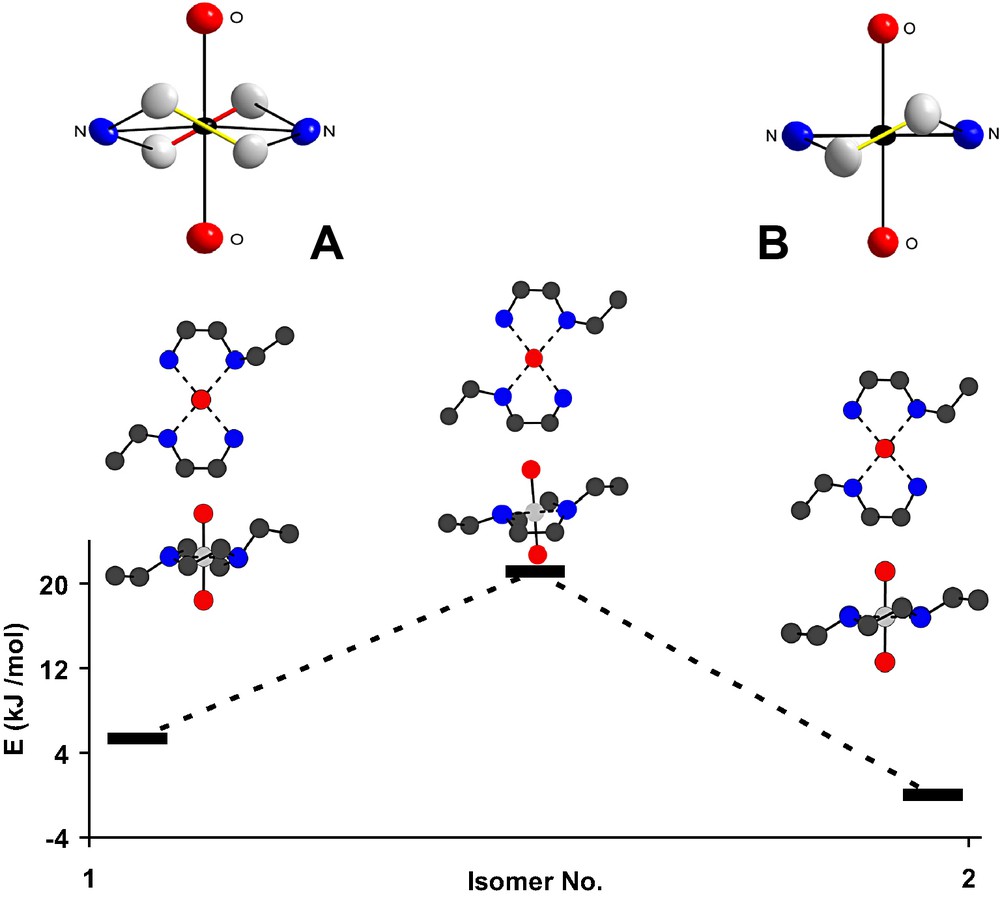
Rotation barrier between two trans-[ReO2(Eten)2]+ cations: staggered (1 in Table 4 and Fig. 5) [Insert A: (δδ + λλ)] and eclipsed (2 in Table 4 and Fig. 5) [Insert B: (δλ)] configurations [via intermediate higher-energy state].
The literature indicates that, for 46 structures evaluated from the Cambridge Structural Database containing ethylenediamine (39 entries), Meen (Meen = N-methylethylenediamine) (six entries) and Eten (one entry), only eight crystallized in a staggered mode, which is less that 20% [27]. It therefore seems that in the solid state there is a preference for this phenomenon, i.e. a ‘self assembly’ in favor of the eclipsed conformation.
This issue has specifically been addressed by Bernal et al. [30] who found that the [(λδ)-Co(en)2(NO2)2]+ cation is thermodynamically preferred over the formation of the [(δδ)-Co(en)2(NO2)2]+ or [(λλ)-Co(en)2(NO2)2]+ cations by a factor of Rln2. The barrier of inversion for the ethylenediamine ring is however fairly low (i.e. 24.7 kJ mol–1) [31]. Substituting one of the NO2– ligands with NCS– destroys the potential inversion center and in this case the pair of en rings for four different crystal structures either had the (δδ) or (λλ) conformation. It would thus seem as if the crystallization pathway is aware of the nature of the internal molecular symmetry and that the packing is affected by such considerations.
We have evaluated this phenomenon by DFT [33] and found the barrier of inversion from eclipsed to staggered (data illustrated in Fig. 4 and Table 4) in the trans-[ReO2(Eten)2]+ cation to be 21.9 kJ mol–1. The relative energy difference between the eclipsed and staggered conformations (entries 1 and 2 in Table 4) is only 5.5 kJ mol–1, but rotation between these two conformers goes through the higher energy transition state (TS in Table 4) [34].
Correlation between solid-state data and computed DFT optimized gas-phase structures. Ethyl functionalized N atoms of Eten ligand has trans configuration
| Complex a | |||||||||||
| 1 | 2 | 3 | X2 | 4 | 5 | 6 | X1 | 7 b | 8 | TS c | |
| Ethyl orientation → | anti | anti | anti | anti | syn | syn | syn | syn | anti | syn/anti | anti |
| Bond lengths (Å) | |||||||||||
| Re=O | 1.785 | 1.783 | 1.784 | 1.766(3) | 1.782 | 1.783 | 1.783 | 1.764(3) | 1.782 | 1.784 | 1.785 |
| Re–N1 (Et) | 2.217 | 2.218 | 2.218 | 2.186(4) | 2.223 | 2.223 | 2.230 | 2.196(4) | 2.214 | 2.225 | 2.206 |
| 2.221 | 2.218 | 2.218 | 2.186(4) | 2.229 | 2.225 | 2.230 | 2.196(4) | 2.212 | 2.218 | 2.215 | |
| Re–N2 | 2.197 | 2.195 | 2.195 | 2.160(4) | 2.196 | 2.198 | 2.198 | 2.163(3) | 2.202 | 2.196 | 2.190 |
| 2.194 | 2.195 | 2.195 | 2.160(4) | 2.201 | 2.198 | 2.198 | 2.163(3) | 2.201 | 2.198 | 2.194 | |
| Angles (°) | |||||||||||
| N–Re–N (bite) | 80.09 | 79.70 | 79.69 | 79.7(2) | 79.66 | 79.52 | 79.70 | 79.2(2) | 79.86 | 79.66 | 75.79 |
| 79.84 | 79.69 | 79.69 | 79.7(2) | 79.70 | 79.52 | 79.69 | 79.2(2) | 80.05 | 79.60 | 79.46 | |
| N–Re–N (non bite) | 99.79 | 100.30 | 100.31 | 100.3(2) | 99.39 | 100.46 | 100.90 | 100.8(2) | 100.65 | 101.35 | 102.22 |
| 100.27 | 100.32 | 100.31 | 100.3(2) | 101.21 | 100.50 | 100.32 | 100.8(2) | 99.44 | 99.39 | 102.53 | |
| Ethyl torsion | 177.95 | 176.95 | 176.94 | 178.3(5) | 86.15 | 88.44 | 75.26 | 65.7(6) | 163.95 | 177.82 | 173.73 |
| Re–N1-C–C (Et) | 163.37 | 176.91 | 176.95 | 178.3(5) | 68.38 | 88.42 | 74.50 | 65.7(6) | 178.14 | 86.28 | 177.24 |
| ν(Re=O) (cm–1) | 884 | 885 | 885 | 798 | 882 | 886 | 881 | 798 | 886 | 887 | 886 |
| Energy Hartrees) | –767.719 | –767.721 | –767.721 | — | –767.717 | –767.718 | –767.716 | — | –767.718 | –767.720 | –767.712 |
| Relative (kJ mol–1) | 5.55 | 0.00 | 0.006 | — | 9.27 | 6.80 | 12.88 | — | 5.93 | 2.57 | 21.96 |
a Complexes as indicated in Figs. 4 and 5; X1 and X2 denote solid-state structures; referenced to Re1 and Re2, respectively.
b Ethyl functionalized N atoms of Eten ligand has cis configuration.
c TS = transition state for inversion from eclipsed (entry 1) to staggered (entries 2 and 3).
It is evident from Fig. 3 and Table 2 and Table 4 that the largest difference between the two independent trans-[ReO2(Eten)2]+ cations is the orientation of the pendant ethyl groups. The Re1 cation could qualitatively be termed ‘syn’ and Re2 as ‘anti’, based on the orientation of the pendant ethyl group, and manifested by the torsion angles. The torsion angle of the Re–N–C–C (Et) moiety is 65.7(6) and 178.3(5)° for the Re1 and Re2 cations, respectively.
We have expanded the investigation regarding conformer formation by computational methods, and studied a range of orientations of the pendant ethyl groups by DFT calculations.
The energy distribution of the range of isomers of trans-[Re(O)2(Eten)2]+ as obtained from DFT are given in Table 4 [energy values in Table 4 given in Hartrees as well as in kJ mol–1, relative energies assuming lowest energy conformer (entry 2) = 0.0 kJ mol–1], and illustrated in Fig. 5. In addition to the two crystallographic conformers, another set of eight isomers were identified and characterized. The trans orientation (with respect to the functionalized nitrogen donor atoms of Eten) was primarily investigated, as well as the rotamers of these complexes, as shown in Figs. 3 and 4. The cis isomer (two functionalized amine groups cis, entry 7 in Table 4), which has not been observed in the solid state or crystallographically characterized to date, was also included in the theoretical study. It displays a higher energy than the trans conformers.
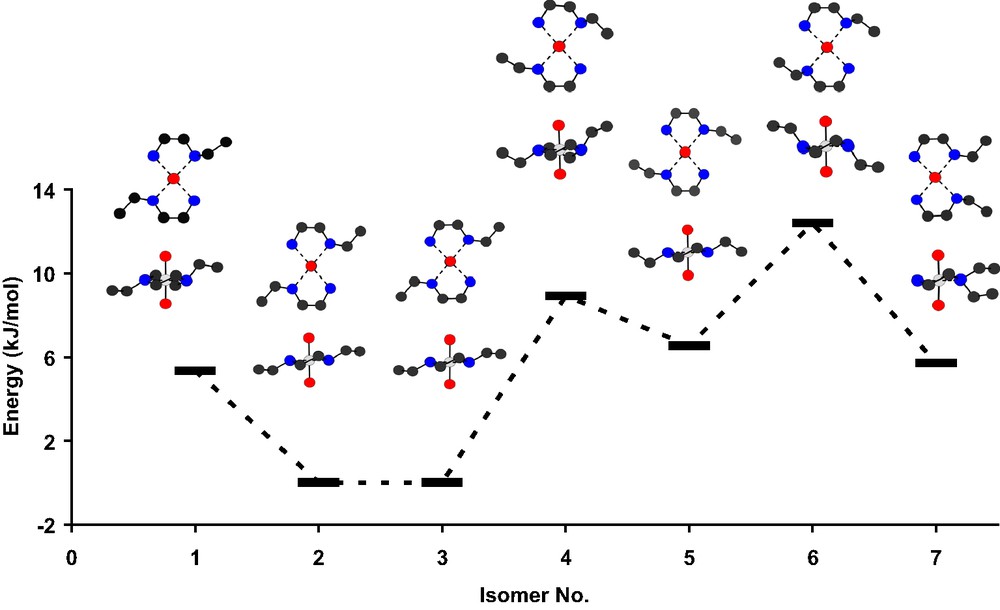
Energy profile of different optimized conformers for the trans-[ReO2(Eten)2]+ cation, including cis-isomer: 7. Conformers 2 and 6 shows excellent correlation with the solid-state X2 and X1 conformers, respectively (see Fig. 6).
There are two distinct ranges of torsion angles for the syn and anti orientations, i.e. 60–90° for the anti (entries 1–3, X2, 7 and TS, Table 4), and ca. 160–180° for the syn (entries 4–6, X1, Table 4) conformers, respectively.
The agreement between the two conformers crystallizing in the solid state and the gas-phase DFT optimized structures is very good (see Table 4 and Fig. 6). The overlay shows only slight differences in the two sets of orientations, indicating that the solid-state structures indeed represent local energy minima. The correlation between computed and crystallographic data are illustrated by the overlay of these structures, as well as by comparing the root mean squared error (RMS error) in each case (see below).
Overlay of crystallographic conformers X1 and X2 with computed conformers 2 and 6, illustrating the excellent structural correlation. The RMS is: (A) X2 vs. 2 = 0.65 Å; (B) X1 vs. 6 = 0.86 Å.
It is interesting to note that the lowest energy isomer (X2, syn conformation), was indeed obtained in the solid state. However, the isomer with the highest energy (X1, anti conformation) was also incorporated during the crystallization process. Thus, the solid-state structure contains a combination of the lower and upper energetic conformers.
Entries 2 and 3, displaying an anti orientation of the pendant ethyl groups, represent an overall minimum with respect to the other conformers/isomers studied, with the difference in energy values between these two conformers being only 6.3 × 10–3 kJ mol–1, with RMS error values of 0.652 Å for entry 2 and X2, and 1.12 Å conformer 3 and X2, respectively. The small difference is attributed to the positioning of the hydrogen atoms on the ethyl pendant groups and the slight distortion of the ethylene carbons connecting the two nitrogen donor atoms. This is indicative of a broadly defined minimum in the region of isomers 2 and 3, although isomer 2 was determined to be the absolute minima in this region of the potential energy space.
In contrast to the above, the second solid-state conformer (X1) and associated isomers, lie on higher energy levels (ca. 6–13 kJ mol–1), with the ethylene moieties folded towards the rhenium metal center, i.e. the syn orientations (manifested in the Re–N–C–C(Et) torsion angles of ca. 70–80°; Table 4 and Fig. 5). Of these, the highest energy conformer is isomer 6, at 12.88 kJ mol–1 which corresponds the best to the crystallographically observed X1, displaying an RMS error of 0.86 Å when overlayed.
Finally, a conformer, termed syn/anti (entry 8, Table 4) was evaluated. In this case, one pendant group is syn (Re–N–C–C(Et) = 86.28°) and the other anti (Re–N–C–C(Et) = 177.82°). A relative energy of 2.60 kJ mol–1, was calculated, which is only slightly larger than that of entries 2 and 3.
In general, the Re=O double bond character, as observed in the average computed bond distance of 1.784 Å, compared to the crystallographic value of 1.764(5) Å, is predicted quite well by the theoretical model. The asymmetric stretching frequency ν(Re=O), for all the computed structures is higher than that experimentally observed. However, it is generally considered that frequency optimization by DFT leads to a ca. 5–10% over estimation [32]. When this is considered, the values of ν(Re=O) in Table 4 approach 800 cm–1, typical of that observed experimentally.
The investigation showed that for this Re(V) system evaluation by solid-state and theoretical methods showed very good agreement, also with regard to IR spectroscopy, enabling a good description of the different conformers. Further studies are underway to include a variation in bidentate functionalization, as well as protonation behavior in these dioxo bis-ethylenediamine d2 systems.
Acknowledgements
Part of this material is based on work supported by the South African National Research Foundation (NRF) under Grant number (GUN 2053397). Any opinion, findings and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NRF.
Financial assistance from the Research Funds of the Free State University and the University of Johannesburg is gratefully acknowledged. The University of the Witwatersrand (Professor D. Levendis; Dr. D. Billing) is thanked for the use of its diffractometer.