

1 Introduction
Transition-metal clusters are a unique class of chemical substances. Not only do they have well-defined molecular structures, they also exhibit interesting and potentially useful properties that are inherent to metal–metal bonded species. As such, there is increasing interest in the studies of metal clusters in order to make use of their well-defined geometry and to fully understand their intrinsically interesting electronic, photophysical, magnetic, and catalytic properties for possible practical applications [1,2].
In this vein, we have been working on the octahedral hexanuclear clusters containing the [Re6(μ3-Se)8]2+ core (Fig. 1) [3,4]. Albeit a recent development in transition metal cluster chemistry, these clusters have received much attention due largely to their well-behaved chemistry and interesting electrochemical and photophysical properties [5,6]. The facile and high-yielding preparation of the starting cluster as the organically soluble [n-Bu4N]3[ReIII5ReIV(μ3-Se)8I6] salt [7] provides synthetic facility not shared by many isostructural systems across the d-block. The cluster core maintains its integrity under most solution-phase conditions, even when undergoing the ca. 1 V ReIII–ReIV redox couple, as suggested by the isolation of the one-electron oxidized halide complex [7]. As well as being structurally sound, the cluster core only allows ligand exchange reactions to occur at the ReIII apices under normal conditions. This fact, combined with the kinetic inertness of the ReIII sites, allows fine control over the substitution chemistry. By carefully controlling the ligand exchange reaction conditions, one can easily prepare a range of isomers and optimize the reaction to enhance the yield of a single one [8,9]. As these cluster complexes are air- and moisture-stable, chromatographic separation of various isomers is straightforward. When the incoming ligand is an inert phosphine, the reaction site becomes protected, only allowing further substitutions to occur at the remaining ReIII apices. The protection process is referred to as ‘site differentiation’ and is a powerful tool for fixing the stereochemistry of the cluster. The ability to fix the cluster stereochemistry turns the site-differentiated species into prefabricated building blocks with shapes commensurate with their stereochemistry.

The structure of the [Re6(μ3-Se)8]2+ cluster core shown also with terminal iodo ligands.
Our efforts have been focused on demonstrating the [Re6(μ3-Se)8]2+ cluster core's utility as a highly generalized synthon for the construction of novel cluster-based molecular and supramolecular arrays [10]. The goals are, however, not only to develop synthetic methodologies, but also to gain electronic understanding of the possibly unique properties of these novel multicluster systems, and eventually, to exploit these properties for possible materials applications.
We report here the synthesis, characterization, and property investigation of a tetrameric array (R4) of the [Re6(μ3-Se)8]2+ clusters featuring a central core of 5,10,15,20-tetra(4-pyridyl)-21H, 23H-porphine supporting four circumjacent [Re6(μ3-Se)8]2+ units (Scheme 1). Metallation of R4 with metal (Co2+, Ni2+, Cu2+, Zn2+) acetates afforded the corresponding cluster-studded metalloporphyrins (R4–Co, R4–Ni, R4–Cu, and R4–Zn). Cluster-based oxidation events have been observed for R4 and each of the metallated derivatives. The observation of a single cluster-based oxidation event attributable to simultaneous removal of four electrons, one from each of the cluster units, suggested minimal electronic communication between the clusters.
2 Results and discussion
2.1 Synthesis and characterization
Novel molecular or supramolecular assemblies containing multiple identical redox centers have attracted much recent interest due to their fundamental and practical significance [11]. In this work, our own efforts in utilizing the redox-active [Re6(μ3-Se)8]2+ clusters as building blocks to construct multicluster arrays and the studies of their electronic properties are described.
The synthesis of R4 took advantage of the substitutional lability of the coordinated nitrile molecule of [Re6(μ3-Se)8(PEt3)5(CH3CN)](SbF6)2 (R) [8] when reacted with pyridyl-based ligands (Scheme 1) [12]. Thus, reacting stoichiometric amounts of the cluster solvate R and 5,10,15,20-tetra(4-pyridyl)-21H,23H-porphine in chlorobenzene under reflux led to the displacement of the solvent ligand and the concomitant formation of the tetracluster R4. This cluster-studded porphyrin features four units of [Re6(μ3-Se)8(PEt3)5]2+ stationed onto the central pyridyl-based tetratopic ligand via N–Re coordination. It is soluble in dichloromethane, acetonitrile, and other common polar organic solvents, yielding brown-red solutions. Despite numerous attempts, X-ray-quality single crystals have so far evaded us.
Several lines of spectroscopic evidence point to the successful production of R4. Upon formation of R4, the 1H signal of the coordinated nitrile of [Re6(μ3-Se)8(PEt3)5(CH3CN)](SbF6)2 (R) disappears, indicating the displacement of the previously attached solvent molecule. This is accompanied by a new signal corresponding to the α-H of the coordinated pyridyl moieties at 9.72 ppm, which is downfield shifted by 0.67 ppm from that of the free ligand. There is also a small but noticeable upfield shift of the β-H of the pyridyl moiety from 8.13 to 8.18 ppm. The pyrrole protons are slightly shifted from 8.86 to 9.04 ppm. There is a peak at –3.00 ppm that is diagnostic of the unmetallated porphyrin due to the two protons associated with the interior nitrogens. The simplicity of the 1H NMR spectrum is consistent with the anticipated symmetric structure. Additional evidence supporting the formation of R4 comes from the unsophisticated 31P NMR spectrum of R4: Two resonances in a 4:1 relative ratio consistent with the stereochemistry of a pentaphosphine-substituted cluster core are shown at –23.6 and –27.6 ppm. R4 was further characterized by MALDI-TOF mass spectrometry: A series of peaks corresponding to the loss of 4, 5, 6, and 7 counter-ions are observed at 2729.4, 2137.9, 1742.5, and 1459.2 a.m.u, respectively. It is clear that the cationic tetracluster maintains its structural integrity under the experimental conditions of the mass spectrometry. Finally, the composition of R4 was confirmed by satisfactory elemental analyses (CHN).
The cluster-studded metalloporphyrins (R4–M; M = Co2+, Ni2+, Cu2+, Zn2+) were prepared via metallation of the prefabricated R4 (Scheme 1). In a representative synthesis, R4 and an excess of metal acetate was dissolved in dichloromethane and stirred under reflux. The resulting mixture contained the metallated product as well as unreacted metal acetate. The solvent was removed, and the product was extracted into dichloromethane. The excess metal salt was removed by repeated washing with water.
The desired metal complexes could be formally synthesized by coupling the metallated tetrpyridylporphyrins with the cluster solvate R. However, this alternative route led only to mixtures containing both the metallated and unmetallated tetracluster porphyrins. Purification by recrystallization or extraction was unsuccessful.
The complexes are soluble in dichloromethane, acetonitrile, and other common polar organic solvents to yield purple-red solutions. 1H and 31P NMR and electronic absorption spectroscopy are in agreement with the proposed structure. The effect of metallation upon the spectra is small. For example, in the 1H NMR of R4–Ni, the α-H of the pyridyl moiety shifts the least from 9.72 to 9.82 ppm. The pyrrole protons and the β-H of the pyridyl have more noticeable shifts of 0.19 and 0.27 ppm, respectively. The most convincing evidence of successful metallation is the disappearance of the resonance at –3.00 ppm, which corresponds to the protons bound to the interior nitrogens in R4. The 31P NMR has two resonances at –24.3 and –27.6 ppm in a 4:1 ratio, indicating that the pentasubstituted cluster motif is present and remains intact. UV–vis spectroscopic studies of porphyrins are diagnostic of both the porphyrin type and the nature of the metal ions, and can be used reliably as evidence for metallation. Details of such studies will be discussed in a separate section below.
Insertion of Cu2+ was achieved in an analogous manner to that of R4–Ni. Upon metallation of the tetracluster porphyrin, dramatic changes to the 1H NMR spectrum occurred. There are three important observations: (1) The peak at –3.00 ppm is no longer present, suggesting that the copper ion has replaced the two protons; (2) the upfield resonances are noticeably broader and less well-defined; and (3) the original resonances in the aromatic region are not visible and only a broad resonance at 9.30 ppm is present. The last two observations are probably due to the influence of the paramagnetic species Cu2+. Little change to the 31P NMR spectra was observed upon metallation, however. There are two slightly broadened resonances at –24.9 and –28.8 ppm in a 4:1 ratio, mandated by the stereochemistry of a pentaphosphine-substituted cluster.
The tetracluster cobalt porphyrin complex R4–Co can be synthesized if cobalt acetate and R4 are refluxed in dichloromethane. As with the copper complex, the 1H NMR of R4–Co is significantly different from that of its unmetallated parent. Again, this can be ascribed to the influence of the paramagnetic Co2+ ion. Disappearance of the original upfield proton signals corroborates the insertion of the metal and the complete displacement of the interior protons. The signals due to the triethylphosphine ligands are broadened, but most interesting are again the signals in the aromatic region. The well-defined resonances associated with the pyridyl and pyrrole protons are replaced by three new resonances, but only one of them is clearly visible at 10.32 ppm. The other two are very broad, at approximately 9.0 and 12.5 ppm, respectively. The 31P NMR spectrum shows two characteristic signals at –25.4 and –28.7 ppm in a 4:1 ratio.
The Zn2+ ion was incorporated into the tetracluster array in a similar fashion. However, when dissolved in dichloromethane, the resulting solution is red-green in color, distinctly different from the purple-red color displayed by the solutions of its Cu2+, Co2+, or Ni2+ cognate in the same solvent. The 1H NMR spectrum clearly indicates the insertion of Zn2+ because the resonance of the protons bound to the interior nitrogen at –3.00 ppm disappears. The 31P NMR has two resonances at –19.3 and –22.6 ppm in a 4:1 ratio, in agreement with the pentasubstituted stereochemistry.
2.2 Electronic spectroscopic studies
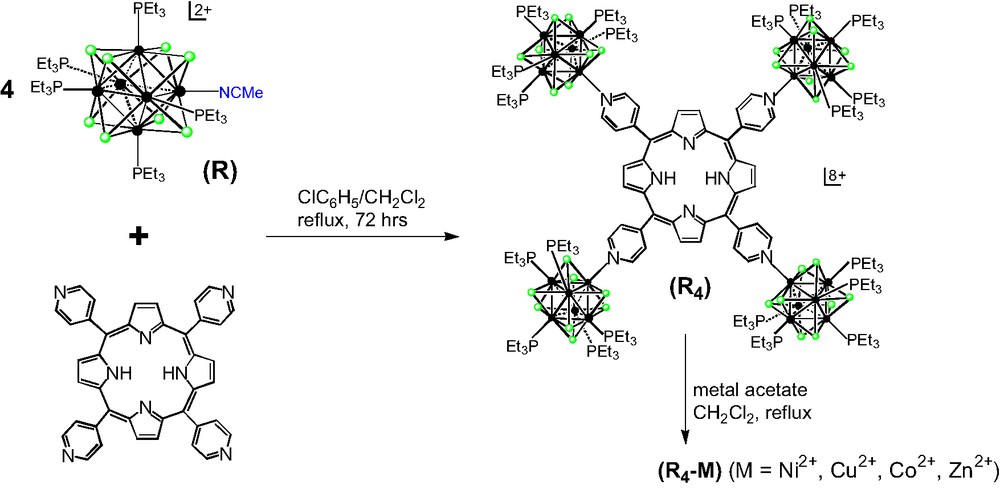
UV–vis spectra of 5,10,15,20-tetra(4-pyridyl)-21H,23H-porphine and R4 are shown in Fig. 2. The electronic absorption data for R4 at 295 K are collected in Table 1. The absorptions of the porphyrin moiety are usual: An intense Soret or B band at 426 nm and weak Q bands at 517, 553, 589, and 644 nm were observed. The B and Q bands both arise from π–π* transitions and can be explained by considering the Gouterman four-orbital model [13]. The absorption spectrum of the tetracluster R4 shows, in addition to the porphyrin Soret and Q bands, poorly defined broad absorptions between 230 and 300 nm that are due to ligand-to-cluster charge transfer [7]. Upon coordination of the clusters, the porphyrin-based absorption peaks are red shifted. This clearly shows that significant interactions exist between these building blocks, but the fundamental properties of both components remain intact upon formation of the array.
UV–vis spectra of 5,10,15,20-tetra(4-pyridyl)-21H,23H-porphine (blue) and R4 (red).
Electronic absorption data of R4
| Complex | Wavelength (nm) | Absorbance | Concentration (mol l–1) | ɛ (l mol–1 cm–1) |
| R4 | 644 | 0.00968 | 1.85 × 10–6 | 5128.2 |
| 589 | 0.01917 | 10 333.9 | ||
| 553 | 0.03000 | 16 172.1 | ||
| 517 | 0.05381 | 29 007.3 | ||
| 426 | 0.85280 | 45 9718.1 |
Metallation of the porphyrin core causes changes to the symmetry of the molecule, and therefore, the π–π* transition. The effects can be seen in the alteration of the Q-band absorbance pattern, and therefore, can be used for identification of specific metallated porphyrin species. As discussed above, the addition of four metal clusters only shifts the electronic absorptions of the porphyrin moiety, but not the configurational interactions. It is therefore reasonable to expect similar electronic spectra of the cluster-modified metalloporphyrins and their unmodified parent.
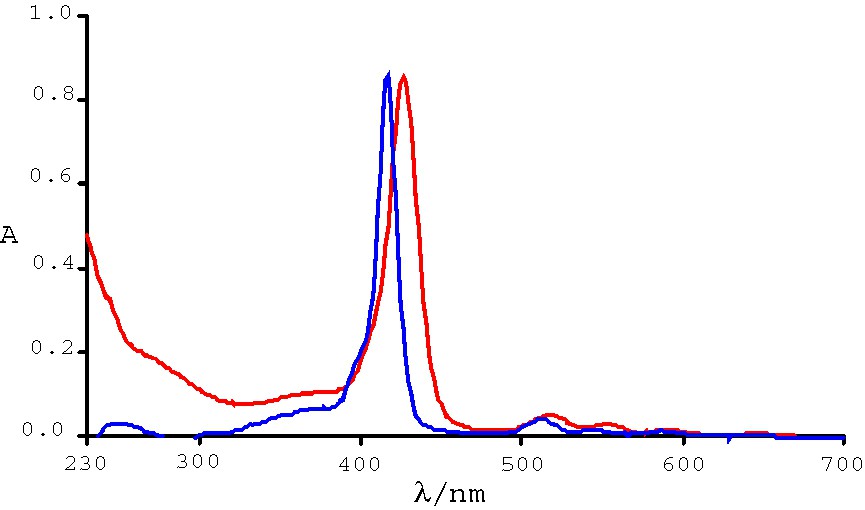
The electronic spectra of R4–Ni, R4–Cu, and R4–Co are shown in Fig. 3, and the absorption data are summarized in Table 2. Upon metallation the Q-band changes significantly: The unmetallated porphyrin shows four Q-band peaks. In contrast, only one Q-band is visible for its Ni2+, Cu2+, or Co2+ complexes. Complete metallation of the porphyrin core is thus inferred.

UV–vis spectra of R4–Ni (blue), R4–Cu (red), and R4–Co (green).
Electronic absorption data of R4–Ni, R4–Cu, R4–Cu, and R4–Zn
| Complex | Wavelength (nm) | Absorbance | Concentration (mol l–1) | ɛ (l mol–1 cm–1) |
| R4–Ni | 530 | 0.0654 | 2.51 × 10–6 | 25977.3 |
| 422 | 0.7478 | 297031.4 | ||
| R4–Co | 544 | 0.0567 | 2.51 × 10–6 | 30159.5 |
| 422 | 0.7770 | 413776.5 | ||
| R4–Cu | 535 | 0.0588 | 2.51 × 10–6 | 23426.3 |
| 426 | 0.5530 | 220318.7 | ||
| R4–Zn | 617 | 0.0335 | 2.51 × 10–6 | 13313.9 |
| 568 | 0.0529 | 21024.0 | ||
| 443 | 0.7205 | 286348.5 |
The electronic spectrum of the non-cluster modified zinc porphyrin (Fig. 4) has only one Q-band at 559 nm. However, in the spectrum of R4–Zn, not only is there a slightly red-shifted Q-band at 569 nm, but a new absorption at 617 nm is also observed. The appearance of this new absorption greatly affects the color observed. Furthermore, upon dissolution in solvents of different polarity, the color of the resulting solution varies significantly. To explore the solvatochromic properties of this system, the electronic absorptions of R4–Zn in organic solvents of varying polarity were studied and the data are collected in Table 3. Similar to other zinc metalloporphyrins, the band position of R4–Zn exhibits a pronounced solvent dependence. The spectral shift of the intense Soret absorption is the easiest to follow. This band can shift up to 14 nm, depending on the solvent employed.
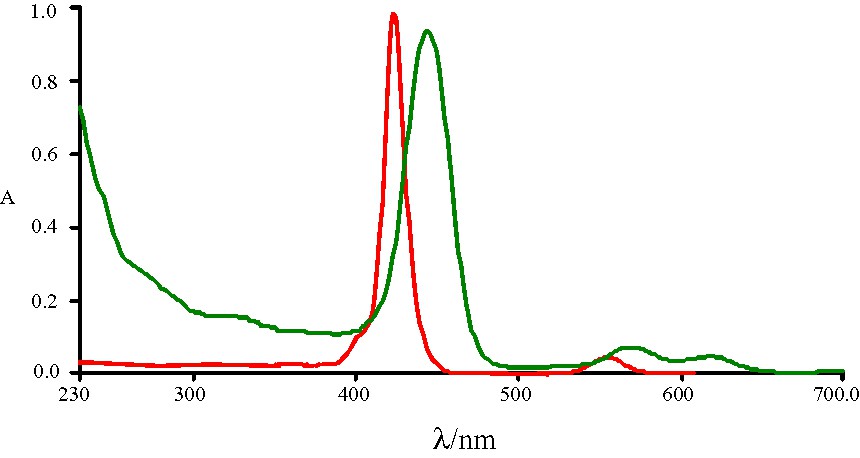
UV–vis spectra of Zn2+ complex of 5,10,15,20-tetra(4-pyridyl)-21H,23H-porphine (red) and R4–Zn (green).
Electronic absorption data of R4–Zn in various solvents
| Solvent | Soret λ (nm) | Q-band 1 λ (nm) | Q-band 2 λ (nm) |
| Acetone | 451.89 | 563.98 | 609.57 |
| Acetonitrile | 441.81 | 562.72 | 606.55 |
| Chloroform | 451.39 | 570.28 | 611.08 |
| Dichloroethane | 440.50 | 562.72 | 605.04 |
| Dichloromethane | 440.30 | 558.19 | 598.99 |
| Dimethylformamide | 453.90 | 574.81 | 623.17 |
| Dimehtyl sulfoxide | 444.84 | 565.74 | 608.06 |
| Nitromethane | 440.83 | 562.72 | 605.04 |
| Tetrahydrofuran | 447.86 | 567.25 | 609.57 |
Many attempts have been made to correlate various experimental parameters with the Soret shifts. But there is really no general correlation between the red shift and dielectric constant [14], Drago's parameters [15], or Gutmann's donor numbers [16]. The behavior of the present porphyrin system is similar to literature observations [14–16]; when a limited number of solvents were used, a correlation with the parameters previously mentioned could be obtained. However, when a larger number of solvents are incorporated in the study, there is no general correlation observed. It was postulated that the limited data set reflects a minor trend of a much more complex phenomenon. Other analyses based upon combinations of parameters fit better the observed shifts [17,18]. However, if the parameters include contributions from many effects, the significance of such a correlation is diminished. Clearly, more data are required before any specific contribution to the spectral shifts can be understood.
2.3 Electrochemical studies
Cluster oxidation potentials for the compounds under the present study are presented in Table 4. A dicluster (R2) bridged by 4,4′-dipyridyl [12] and a tricluster (R3) bridged by 2,4,6-tri(4-pyridyl)-1,3,5-triazine [19] are included in the study in order to correlate the number of electrons involved in the redox events to the number of clusters in the molecular arrays. The potentials represent the half-wave potential from cyclic voltammetry (CV) experiments in acetonitrile at a Pt disc electrode (100 mV s–1), referenced to the ferrocenium/ferrocene (Fc+/Fc) redox couple (E° = 0.46 V vs. SCE; 0.70 V vs. NHE). Oxidation potentials are not significantly changed (ca. 0.70 V) in going from the free cluster solvate [Re6(μ3-Se)8(PEt3)5(CH3CN)](SbF6)2 (R) to assemblies of up to 4 clusters per molecules (i.e. the R4 series), and all materials give highly reversible CVs on this time scale with ΔEp values of ca. 70 mV, which is comparable to those observed for ferrocene in this cell, under the same experimental conditions.
Oxidation potentials and number of electrons transferred for cluster monomer to tetramers
Electrochemical measurements were made at 293 K in acetonitrile solutions containing subject compounds at ~1 mM effective cluster concentration, in addition to 0.3 mM TBHFP supporting electrolyte
| Compound | EOx1 vs. Fc+/Fc (V) | Electrode volume (μl) | Q/C (μC mM–1) | n |
| R | 0.69 | 0.466 | 40 | 0.9 |
| R2 | 0.72 | 0.373 | 79 | 2.2 |
| R3 | 0.71 | 0.415 | 119 | 3.0 |
| R4 | 0.68 | 0.415 | 153 | 3.8 |
| R4–Zn | 0.71 | 0.466 | 184 | 4.1 |
| R4–Ni | 0.72 | 0.373 | 134 | 3.7 |
| R4–Cu | 0.70 | 0.373 | 137 | 3.8 |
| R4–Co | 0.71 | 0.394 | 142 | 3.7 |
In order to obtain high-resolution and high-signal-to-background voltammograms and to extract absolute coulometric information for these materials, a thin-layer cell electrode (TLE) was developed (see Section 4 for details) [20] and utilized to determine whether these materials exhibit coupled or uncoupled oxidations.
Thin-layer cyclic voltammetry (TLCV) and coulometry measurements were made in the same solution cell as the standard voltammetric measurements, exhaustively oxidizing the analytes in the confined volume defined by the TLE. At sufficiently slow scan rates (v, developed below), the current measured in a voltammetric experiment has no dependence on mass transfer in the cell, and it is therefore not necessary to know the diffusion coefficient (D0) of the redox species in order to determine the number of electrons transferred per redox event (n) [21]. While the peak current in such a voltammogram is directly proportional to v, the total amount of charge passed (Q) in exhaustively oxidizing/reducing the contents of such a thin-layer is independent of v and can be related to n by the following formalism(Eq. 1)
TLCVs (1 mV s–1) of selected subject compounds (R, R2, R3, and R4–Zn), containing between one and four clusters per molecule, are presented in Fig. 5. The current axis in this plot has been normalized to the number of molecules in the confined volume, and on this scale, the peak areas are proportional to the amount of charge passed (number of electrons) per molecule. Q/C values were generally calculated by integrating the anodic peak in the original current vs. voltage plots and dividing by scan rate and concentration. These values are listed in Table 4 for all materials, along with the calibrated cell volume for that measurement. The number of electrons transferred per molecule for the cluster oxidation event, n, was then calculated using Eq. 1 and is also reported in Table 4. It was determined that for every compound studied, all clusters on a given molecule oxidize at the same potential, without influencing each other (i.e. these are ‘non-interacting’ redox couples), yielding a measured value of n that is approximately equivalent to the number of clusters per molecule. Based on this determination of n, diffusion coefficients were calculated to be approximately 5 × 10–7 (cm2 s–1) from standard voltammograms for these materials, placing this determination (at v = 1 mV s–1) within the valid working range of the TLE.

Normalized thin-layer cyclic voltammograms (1 mV s–1) for representative monoclusters to tetraclusters.
For the R4 series, R4 and R4–Co give nearly identical CVs to that shown for R4–Zn in Fig. 5. R4–Ni and R4–Cu also exhibit a 4-electron cluster oxidation, but additional oxidations are observed in this potential window. These extra peaks are attributable to core metal oxidations, which are not reversible on this very slow (1 mV s–1) time scale. The return sweep (cathodic or top peak) of the cluster oxidation was used for cluster coulometry measurements on these materials (4 e–), as there was no interference from the irreversible core metal oxidation. ΔEp values (<100 mV) for all materials are comparable to those of ferrocene and are the result of uncompensated cell resistance within the TLE capillary.
At a faster time scale (10 mV s–1), where the voltammetric response resides between thin-layer and diffusion-limited behavior for molecules of this size, the core metal oxidations observed for R4–Cu and R4–Ni are reversible, as shown in Fig. 6. At this sweep rate, absolute coulometric measurements are invalid, yet superior resolution and Faradaic current response is achieved over a typical Pt disc electrode, where core metal oxidations for R4–Cu were not detectable. Relative peak areas for separate redox processes of the same molecule, however, are still expected to be proportional to the number of electrons passed per redox event. Using peak fitting, area ratios were determined to be ca. 1:1:4 (core/core/cluster) for R4–Cu and 1:2 (core/cluster) for R4–Ni. Based on the previous determination of a 4-electron oxidation for the cluster peak of both species at slow scan rates, the stoichiometry for core metal oxidation can be inferred: two 1-electron oxidations are obtained for R4–Cu (CuII → CuIII → CuIV) and one 2-electron oxidation is obtained for R4–Ni (NiII → NiIV), which is in agreement with other coulometric studies on Cu and Ni-cored metalloporphyrins [22]. It appears that the multiple cluster oxidations on these molecules are not influenced by each other or by metal core oxidations, and remain uncoupled. Additionally, these R4-series compounds were observed to have varying reduction energies, attributable to the porphyrin core or metal, which have no influence on the cluster oxidation.

‘Fast’ thin-layer cyclic voltammograms for R4–Cu and R4–Ni; weep rate (10 mV s–1).
3 Conclusions
In summary, a star-shaped tetrameric array of the [Re6(μ3-Se8)]2+ clusters supported by a central porphyrin scaffold was prepared. Four metal complexes of this cluster-studded porphyrin were also synthesized successful. Interestingly, for both the porphyrin and each of its metal complexes, non-interaction between the clusters has been inferred from the observation of only a single redox wave at a potential that is essentially identical to that of a monocluster. Coulometry confirmed the electron counts of 4, one from each of the peripheral cluster units. Perhaps the cluster is predominantly capable of sustaining the charge associated with the oxidized state, without substantially polarizing neighboring clusters, even when conjugative linking groups are present.
4 Experimental
4.1 Materials and methods
The cluster solvate [Re6(μ3-Se)8(PEt3)5(CH3CN)](SbF6)2 was prepared according to published procedures [8]. 5,10,15,20-Tetra(4-pyridyl)-21H, 23H-porphine, (n-Bu4N)PF6, and other reagents were of commercial origin and were used as received. NMR (1H and 31P) spectra were recorded on a Varian Unity 300 spectrometer in CD2Cl2 solutions. Chemical shifts of 31P NMR spectra were referenced to 85% H3PO4 (0.0 ppm, with negative values meaning upfield). NMR resonance multiplicities are designated: s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Mass spectrometry data were collected on a Bruker Reflex III MALDI-TOF instrument. A conventional N2 laser (337 nm) was used in the experiments with variable attenuation (typical range: 40–60%). Dithranol was used as a matrix, which was dissolved in CH2Cl2 (0.1 M solution). This matrix solution of dithranol was mixed with a 0.1 M CH2Cl2 solution of the sample in a 5:1 ratio, and 1 μl of this mixed solution was deposited on the MALDI plate. Electronic absorption spectra in dichloromethane solutions were recorded on a Perkin Elmer Lambda 10 spectrophotometer. Elemental analyses (CHN) were performed by Desert Analytics Laboratory, Tucson, AZ.
4.2 Synthesis of R4
A mixture of [Re6(μ3-Se)8(PEt3)5(CH3CN)](SbF6)2 (R, 274 mg, 0.0962 mmol) and 5,10,15,20-tetra(4-pyridyl)-21H, 23H-porphine (10.0 mg, 0.0323 mmol) in 30 ml of chlorobenzene was stirred under reflux for 72 h. An orange-red residue was obtained upon removal of solvent. The residue was dissolved in 5 ml of dichloromethane, and the solution was triturated with ether to give an orange-colored powder. The product was obtained as dark red crystals after recrystallization from ether and dichloromethane. Yield: 157 mg (82.0%). 1H NMR: –3.00(s), 1.05–1.30(m), 2.10–2.40(m), 8.18(d), 9.04(s), 9.72(d). 31P NMR: –23.6(s), –27.6(s). MS: m/z 2729.4 [M-4SbF6–]4+, 2137.9 [M-5SbF6–]5+, 1742.5 [M-6SbF6–]6+, 1459.2 [M-7SbF6–]7+. Anal. Calc. for Re24Se32P20N8C160H324Sb8F48: C, 16.19; H, 2.73; N, 0.94. Found: C, 16.50; H, 2.75; N, 0.98.
4.3 Synthesis of R4–Ni
A mixture of R4 (50.0 mg, 0.004 mmol) and Ni(C2H3O2)2·4 H2O (60.0 mg, 0.241 mmol) in 30 ml of chloroform was stirred under reflux for 48 h. The red residue obtained upon removal of the solvent was extracted using dichloromethane and water. The organic phase was dried over MgSO4. A purple–red residue was obtained upon removal of the solvent. Recrystallization from ether and dichloromethane afforded purple-red crystals as the product. Yield: 45 .0 mg (90.3%). 1H NMR: 1.00–1.30(m), 2.00–2.40(m), 7.91(d), 8.85(s), 9.62(d). 31P NMR: –19.5(s), –22.8(s). Anal. Calc. for Re24Se32P20N8C160H324NiSb8F48: C, 16.13; H, 2.72; N, 0.94. Found: C, 16.48; H, 2.89; N, 1.10.
4.4 Synthesis of R4–Cu
R4–Cu was obtained as a purple powder in a similar manner to R4–Ni except that Cu(C2H3O2)2·H2O was used in place of Ni(C2H3O2)2·4 H2O. Yield: 0.046 g (92%). 1H NMR: 0.80–1.10(m), 1.85–2.25(m), 9.30(bs). 31P NMR: –24.9(s), –28.3(s). Anal. Calc. for Re24Se32P20N8C160H324CuSb8F48: C, 16.12; H, 2.72; N, 0.94. Found: C, 16.42; H, 2.90; N, 1.07.
4.5 Synthesis of R4–Co
R4–Co was obtained as a purple powder in a similar manner to R4–Ni except that Co(C2H3O2)2∙4H2O was used in place of Ni(C2H3O2)2·4 H2O. Yield: 0.046 g (92%). 1H NMR: 0.55–1.33(m, 180), 1.85–2.50(m, 120), 9.03(bs), 10.32(bs), 12.49(bs). 31P NMR: –25.4(s), –28.7(s). Anal. Calc. for Re24Se32P20N8C160H324CoSb8F48: C, 16.13; H, 2.72; N, 0.94. Found: C, 16.18; H, 2.91; N, 1.07.
4.6 Synthesis of R4–Zn
R4–Zn was obtained as a green powder in a similar manner to R4–Ni except that Zn(C2H3O2)2∙2H2O was used in place of Ni(C2H3O2)2·4 H2O. Yield: 0.047 g (94%). 1H NMR: 1.00–1.30(m), 2.04–2.40(m), 8.09(d), 8.95(s), 9.67(d). 31P NMR: –19.3(s), –22.6(s). Anal. Calc. for Re24Se32P20N8C160H324ZnSb8F48: C, 16.12; H, 2.72; N, 0.94. Found: C, 16.47; H, 2.69; N, 1.11.
4.7 Cyclic voltammetry
CVs were acquired with an EG&G Instruments 283 potentiostat, using a 500 μm diameter Pt working electrode, a freshly prepared Ag/AgCl pseudoreference electrode, and a Pt counter electrode, in N2-degassed solutions containing (n-Bu4N)PF6 as supporting electrolyte. All solvents were purified by distillation and stored over activated alumina to remove trace water. Prior to preparing analyte solutions, the distilled solvent was run through a fresh column of activated alumina. Unless otherwise noted, an analyte concentration of ca. 1 mM, a scan rate (v) of 100 mV s–1, and an electrolyte concentration of 0.1 M was used. A custom glass solution cell with fritted openings was used to stabilize the electrodes and minimize permeation of oxygen. The working electrode was cleaned between each experiment by polishing with 0.3 μm alumina paste for 1 min, followed by copious solvent rinses. The reference electrode was prepared by plating a thin-layer of AgCl onto a Ag wire from a 1 M KCl solution. After each voltammetric experiment, ferrocene was added (ca. 1 mM), an additional voltammogram was recorded, and the potential axis was calibrated against the formal potential for the Fc+/Fc redox couple. Reported redox potentials represent the half-wave potential in these voltammograms.
4.8 Thin-layer cyclic voltammetry/coulometry
A thin-layer cell electrode (TLE) was designed and fabricated in order to perform exhaustive electrolysis on analyte solutions, yielding coulometric information for the relevant redox event. A schematic drawing of this electrode is presented in Fig. 7 [20]. The electrode was designed such that these measurements could be made using the above setup for standard cyclic voltammetry, where the working electrode is simply replaced with the TLE. The entire electrode assembly was placed in the solution cell, and the distal end was submerged 2–5 mm into the analyte solution, causing the solution to reproducibly fill the entire cavity created between the Pt wire and the small capillary due to capillary action. Relatively large electrolyte concentrations (0.3 M) were necessary to minimize the potential drop in the TLE cavity. TLCVs were collected at 1 mV s (where there was insignificant diffusion dependence on the current), and the cell volume was first calibrated using a 1 mM standard solution of ferrocene in acetonitrile. Following the confirmation of a one-electron oxidation for the free cluster monomer R, standard solutions of R were used for volume calibration prior to analyzing multicluster molecules. Solvent background (minimal) was subtracted from all charge measurements.

Schematic of thin-layer cell electrode.
Acknowledgments
We wish to acknowledge Research Corporation and University of Arizona for financial support of this work.