

1 Introduction
Even if progress in protein structure determination has been tremendous over the last years, still large classes of proteins cannot be investigated using liquid state NMR or X-ray crystallography, either because the proteins cannot be crystallized to a sufficient diffraction quality for X-ray diffraction, or they cannot be brought into a sufficiently concentrated solution, or are too large for liquid-state NMR. For example, even though membrane proteins represent 30% of the proteome, relatively little is known about the structure of these proteins, because of their poor capacity to yield diffracting crystals. Therefore there is a considerable interest in the development of methods for protein structure determination, which do not have these limitations. High-resolution solid-state NMR (SSNMR) is a very promising technique in this respect, and has been subject during recent years to intense development by several groups all over the world. We currently assist the progression from studies using selective isotope labeling of the proteins under investigation, to the observation of uniformly isotope-enriched samples, which allow extracting structural information directly. This movement has been made possible by decisive advancements in NMR spectrometer hardware and methodology, as well as major progress in the preparation of uniformly labeled protein samples.
This review highlights the recent advances in protein high-resolution SSNMR, comprising resonance assignments and structural analysis of fully labeled proteins. Recent developments concerning protein dynamics, and proteins as interaction partners, as well as methodological developments made on protein samples, will be illustrated.
2 Protein high-resolution magic angle spinning (MAS) NMR
Protein MAS SSNMR has proven its capability of answering pertinent question in biology for more than a decade (see for example [1–7]). Most of these studies, however, addressed specific questions, which could be answered by looking at one specific site in the protein, measuring one decisive inter-atomic distance, or looking at one specific interaction predicted by structural models. Multiple site analysis was long time hampered by a lack of spectral resolution, mainly caused by insufficient averaging of the strong dipolar couplings present in solids. Important breakthroughs in instrumentation, e.g. high field strength, decoupling powers, spinning speeds, and advances on the methodological side, e.g. pulse sequence design, opened the way to high resolution. This enables site-specific observation even for larger systems, corresponding to a heap from some few to several hundred spins. On the biological side, this progress was accompanied by a major advancement: the better understanding of how to produce and isotopically label large amounts of protein, e.g., on the mg scale. As a result, starting only a few years ago, several SSNMR spectroscopy laboratories have become engaged in solving the complete structures of biological macromolecules using high-resolution methods based on MAS. These efforts typically involve structurally homogeneous samples, and utilize recently developed pulse sequences for the sequential correlation of resonances, the detection of tertiary contacts and the characterization of torsion angles (for a review, see reference [8]).
3 Sample preparation
Immobilized globular proteins are ideal candidates to establish and improve the necessary NMR techniques. One first important step was thus the possibility to prepare homogenous samples of these proteins, and demonstrate that current NMR methodology is able to produce spectra amenable to site-specific assignment. Pioneering work by Cole et al. [9], as well as Creuzet et al. [10], illustrated early-on that excellent NMR line widths can be obtained for microcrystalline or precipitated hydrated globular systems. More recently, several methods were tested to produce solid protein samples, including liophylization, precipitation and crystallization. Micro- or nanocrystals have shown to yield the best-resolved spectra so far. Currently, sample preparation for SSNMR studies of globular proteins is done by micro- or nanocrystallization of the protein using classic precipitants, including PEG and MPD [11–17]. Crystallization is either induced naturally, or accelerated by dehydration, typically using a concentrated salt solution in the crystallization reservoir [13], or a speedvac to concentrate the solution [14]. In general, high protein concentration is used to obtain quantitative precipitation. Several groups demonstrated in the last several years that high-resolution solid-state MAS spectra can be obtained using this type of samples [11–13,18]. Care has however to be taken as to the salt content of the precipitant and buffer, as quality factors of the probe suffer from high salt conditions.
Crystalline proteins have proven to yield well-resolved spectra in all cases; it has however been demonstrated recently that crystallinity is not a prerequisite for obtaining high-resolution spectra [19–21]. The exact relationship between the physical state of the protein and the observed line widths however remains only partly understood.
4 Sequential assignments of uniformly labeled proteins
Assignment of the spectra is the first compulsory step towards structure and dynamics analyses of a protein by NMR methods. Several groups demonstrated that high-resolution multidimensional SSNMR methods can be used to correlate many backbone and side chain chemical shifts to obtain sequential assignments for small proteins of up to about 100 amino acids. Partial assignments have been obtained for ubiquitin [18] and BPTI [12]. Nearly complete assignments have followed for the SH3 domain of α-spectrin, Crh [13], ubiquitin [15,16], thioredoxin [17], and KTX [19]. For the uniformly labeled HET-s prion protein, partial assignments were published recently [20]. Assignments of these proteins were obtained using multidimensional experiments originally developed for pairs of isolated spins, but which showed to perform equally well on uniformly labeled proteins. MAS is used together with high power proton decoupling to achieve narrow lines, in the order of 1 ppm or less. Two-dimensional carbon–carbon correlation spectra have sufficient resolution to identify spin systems; typical experiments include proton driven spin diffusion [22], RFDR [23], DREAM [24] or RAD [25]. Double quantum experiments [26–28] as well as experiments using J-couplings [29–32] allow us to select for one-bond transfers. Nitrogen–carbon correlations can be established using selective variants of heteronuclear chemical shift correlation experiments [33], in order to minimize magnetization losses.
Figs. 1 and 2 illustrate this for the 10.4-kDa carbon catabolite repression HPr like protein (Crh). Fig. 1 shows the 2D 13C–13C PDSD spectrum used to identify the spin systems; Fig. 2a shows the 15N–13C correlation spectra used for identification of the amino acid 15N chemical shifts, and Fig. 2b the spectrum used for sequential assignments. 3D NCOCA, NCACO, and NCACX experiments allow sequential assignments in a very similar manner to liquid-state NMR, as has recently been shown for ubiquitin [16]. For most model proteins, solution and solid-state chemical shifts did not differ by more than about 1 ppm. Several of the assigned proteins, as SH3, Crh and ubiquitin, now serve as valuable model system for additional SSNMR technique developments, for example for protein structure, dynamics or protein–protein or protein–ligand interactions, as described in the following.
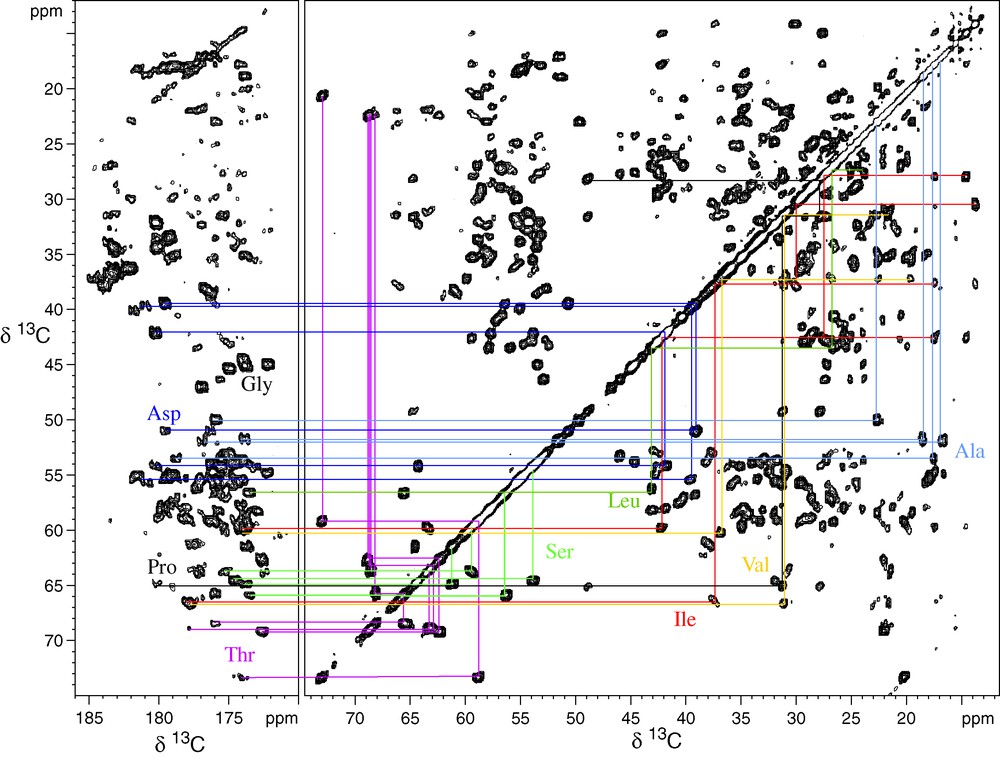
Proton-driven spin diffusion of the microcrystalline Crh protein. An extract of the carbonyl region, as well of the aliphatic correlations are shown. Several spin systems are illustrated. Most spin systems could be assigned using this spectrum [13]. (Reprinted from Böckmann, Lange, Galinier, Luca, Giraud, Juy, Heise, Montserret, Penin and Baldus, J. Biomol. NMR 27 (4) (2003) 327, with kind permission of Springer Science and Business Media).

Heteronuclear correlation spectra of the Crh protein. The spectrum in (a) shows the NCACB correlations used for identification of the nitrogen resonance for each spin system. Spectrum (b) shows the NCOCACB cross signals used for sequential assignments [13]. (Reprinted from Böckmann, Lange, Galinier, Luca, Giraud, Juy, Heise, Montserret, Penin and Baldus, J. Biomol. NMR 27 (4) (2003) 331, with kind permission of Springer Science and Business Media).
5 Structural analysis
Tertiary structure determination is one of the milestones in protein NMR. First structural information can be obtained from chemical shift information. It has been shown for the Crh protein [13] that chemical shift analysis, and the resulting simulated dihedral angles obtained from a TALOS-based statistical analysis, indicate that the microcrystalline arrangement of Crh is similar to the domain-swapped dimeric structure of a single crystal form [34]. This is illustrated in Fig. 3, which compares the predicted angles from the SSNMR chemical shifts to the dihedral angles of the monomer and dimer Crh structure. Another study uses dihedral angles predicted from chemical shifts to determine the conformation of neurotensin bound to its G protein-coupled receptor [35]. Dihedral angles predicted from chemical shifts have also been used in molecular-dynamics-based structure calculation [19]. This illustrates that the assigned spectra are immediately useful for assessing secondary structure.

Structural information obtained from chemical shifts. Shown is the comparison of the predicted dihedral angles using 15N and 13C chemical shift information to the dihedral angles of the monomeric (a) and dimeric (b) form of Crh. It can be seen that the dihedral angles closer fit the dimeric (d) than the monomeric (c) Crh structure [13]. (Reprinted from Böckmann, Lange, Galinier, Luca, Giraud, Juy, Heise, Montserret, Penin and Baldus, J. Biomol. NMR 27 (4) (2003) 337, with kind permission of Springer Science and Business Media).
Aiming at the use of protocols developed in liquid state NMR studies, long-range constraints have to be obtained for structure calculations. Strategic labeling together with simple spin diffusion experiments has recently led to a moderate-resolution structure of the SH3 protein by SSNMR [36,37]. Partial deuteration is another promising approach to obtain long-range constraints [25]. Deuteration is however not always without influence on the resolution of protein spectra (Böckmann et al., accepted in J. Biomol. NMR). Selective recoupling methods have been forwarded as an alternative to selective labeling [38]. It has been shown that the spin system dynamics remain sensitive to the distance of interest in chemical shift selective experiments and can be well reproduced within a quantum-mechanical multiple-spin analysis [39], making it possible to measure long-range constraints in uniformly labeled proteins. Selective transfer from carbonyl to side chain carbons [40] or between nitrogen and carbon spins [41] is another attractive approach for the measurement of long-range constraints. Recently, a set of 3D NMR spectroscopy for structure elucidation of proteins under MAS has been proposed [42]. It relies on the use of proton homonuclear transfer, bracketed by 13C and/or 15N evolution times. This scheme has shown to be able to produce high-resolution spectra connecting carbon or nitrogen spins through-space [43,44], and has already been applied successfully to the identification of the dimer interface of the Crh protein [45]. Most recent work shows that these heteronucleus edited, proton relayed distance measurements can be used for calculations of the 3D structure of solid proteins [19]. This approach is highly attractive, as it can be carried out on a uniformly labeled sample and does not require supplementary block labeled samples.
As already mentioned before, backbone dihedral angles carry structural information as well. In addition to predicting them from chemical shift information, direct determination is possible by the correlations between NH/NH [46] and NH/CH [47] dipolar tensors. Alternatively, relative tensor orientation may be encoded in the evolution of a 2Q two-spin state under the effect of two anisotropic interactions [48–50]. Multiple backbone torsion angles can be measured in fully labeled proteins using 3D versions of these experiments [51].
6 Protein–protein and protein–ligand interactions
Intermolecular contacts often provide the microscopic basis for molecular structure and function, ranging from the definition of macroscopic sample properties in material science applications to the control of cellular processes. In the latter case, protein–protein complexes, oligomerization during protein folding or ligand binding to membranes or membrane receptors exemplify conditions where such interactions can occur in a non-crystalline and insoluble environment. As demonstrated in enzyme–substrate complexes [52], membrane peptides [53–55], and amyloid fibrils [56,57], intermolecular contacts can be readily probed by SSNMR methods. In these studies, mutagenesis, X-ray/NMR structures or other biophysical parameters were used to place specific isotope labels in the molecular region of interest.
Information on entire molecular segments becomes accessible if multiply or uniformly labeled protein variants are studied. Using SSNMR schemes that permit probing of individual dipolar couplings under MAS conditions, the through-space distance between two spins can be subsequently determined. A general NMR strategy was recently introduced to directly study molecular interfaces under MAS conditions [45]. The approach is based on the spectroscopic analysis of mixtures composed of different molecules, uniformly labeled with spin species X or Y (denoted X:Y). Application of an NHHC 2D experiment to an (15N:13C) labeled sample of the Crh domain-swapped dimer revealed a variety of backbone–backbone and backbone–side chain contacts, as illustrated in Fig. 4. The signals in the spectrum all originate from inter-monomer NHHC magnetization transfers. The contacts could be assigned to the residues indicated on the Crh dimer crystal structure [34] in purple and blue. Contacts predicted from the structure but not identified in the spectrum are shown in gray.

2D NHHC spectrum of the 15N:13C labeled Crh dimer. Cross signals origin from inter-monomer proton mediated magnetization transfer. On the structure are reported the observable contacts in purple and blue spheres. Predicted, but not observed contacts are indicated in gray [45]. (Reprinted with permission from J. Am. Chem. Soc. 126 (2003) 14750, 14751. Copyright (2003) American Chemical Society).
An approach to characterize ligand binding to a protein in solid samples has been described recently [58]. It has been shown that binding of a small peptide or drug-like organic molecule leads to changes in the chemical shift of resonances from multiple residues in a uniformly labeled protein that can be monitored to characterize binding. Differential chemical shifts have been used to distinguish between direct protein–ligand contacts and small conformational changes of the protein induced by ligand binding.
7 Protein dynamics
The analysis of molecular dynamics in proteins is one of the key challenges in understanding their structure–function relationships. Well-established solution-state NMR methods currently provide detailed information about local dynamics throughout the protein. For solid proteins, nuclear spin relaxation times provide a direct and unambiguous measure of the presence of molecular dynamics, which cannot be confused with static structural disorder. The link between nuclear relaxation times in solids and motion has been established for decades [59], and several deuterium NMR line shape and relaxation studies [60–62] have been performed on solid-state proteins in the past. Cole and Torchia [63] have also measured nitrogen-15 relaxation times for specific sites in a protein. Widespread multiple site-specific information, crucial to most modern models of protein interactions and function, has only recently be obtained [64] using high-resolution spectra provided by the recent progresses of protein SSNMR. The measurements of nitrogen-15 nuclear longitudinal relaxation rates were reported for a microcrystalline sample of the protein Crh, providing a qualitative description of the site-specific backbone dynamics in the solid state. Heteronuclear 15N–13C correlation experiments were acquired, based on a double cross-polarization technique, and including a spin-lattice relaxation during a variable delay after the 1H–15N CP. The resulting spectra allow the determination of the 15N longitudinal relaxation times of many resolved resonances. Substantial differences (up to a factor 7) in R1 were observed along the backbone, as shown in Fig. 5 (top panel). It was apparent that there is a strong correlation between measured relaxation rates and a simple but coherent preliminary picture of internal mobility, with increased mobility providing faster R1. In particular, increased mobility was found for residues that are not in regular secondary structures, whereas the rates measured for residues in helices or β-sheets showed less mobility. This is illustrated in Fig. 5 on the X-ray crystal structure of the protein, where the different residues are color coded according to their relaxation rates. Relaxation in solid proteins seems thus to correlate to structural features, as already observed for proteins in solution. It is noteworthy for example that the fastest relaxing residue in Crh, Ala 54, undergoes a major dislocation on domain swapping and appears to remain mobile in the dimer.

Relaxation rates determined by 2D 15N–13C correlation spectroscopy including a 15N relaxation time are shown for the resolved residues. On the Crh 3D structure fast relaxation rates are color coded in red, intermediate in yellow, and slow in blue [64]. (Reprinted with permission from J. Am. Chem. Soc. 126 (2003) 11423. Copyright (2003) American Chemical Society).
Dynamic behavior was also measured for the SH3 domain, using 2H–13C correlation spectra. 2H quadrupolar couplings were fitted to side chain motional models, and compared to electron densities deduced from the SH3 crystal structure [65].
8 High-speed MAS
Proteins are not always available in amounts desirable for solid-state MAS nuclear-magnetic resonance spectroscopy. To maximize the signal-to-noise ratio achievable with small amounts of sample, the filling factor must be optimized by using small-diameter MAS rotors. These rotors have the added benefit of allowing higher radio frequency field amplitudes during polarization transfer steps and during decoupling periods as well as allowing higher spinning frequencies. It has long been thought that high speed MAS is not suitable for protein samples, due to sample heating. It has however been shown that microcrystalline proteins are amenable to fast MAS, and that signal-to-noise obtained with the small sample volumes used is adequate for multidimensional heteronuclear spectroscopy [66]. Studies at even higher speeds show that protein high-resolution spectra can be obtained, provided that adequate cooling devices [67] are used (A. Samoson, personal communication).
9 Resolution and sensitivity improvement
One of the principal factors limiting the study of larger proteins by SSNMR remains spectral resolution. In uniformly 13C-labeled compounds such as proteins, the 13C–13C J-couplings constitute a significant contribution to the line width in MAS spectra. The application of spin-state selective and transition-selective polarization transfer to multidimensional SSNMR correlation experiments of 13C-labeled proteins removes the line broadening due to the JCO–Cα spin coupling in both direct and indirect dimensions of a two-dimensional correlation experiment and allows for a nearly twofold improvement in line width [68].
In addition to resolution, an important limitation to the application of SSNMR to more complex, or less concentrated, systems is sensitivity. This limitation is particularly acute for the multidimensional experiments that are the cornerstone of spectral assignment and structure determination. Sensitivity is routinely improved by cross-polarization and by spin decoupling, which concentrates the intensity into a narrow resonance. In many solids, however, decoupling only improves resolution and sensitivity up to a point where the remaining line width is dominated by susceptibility effects or chemical shift distributions. It has been shown however that even once the limiting line width has been reached, decoupling sequences continue to act strongly on the transverse dephasing times that determine the sensitivity of many multidimensional or multipulse experiments [69].
10 Water–protein interactions in solid-state proteins
The sensitivity of NMR to motions on time scales of chemical events also allows the study of interactions between proteins and solvent. Water–protein interactions include interactions with structural or bound water, surface water, or interaction by chemical exchange between protein protons and water protons. NMR has proven to be a powerful tool to study protein/water interactions in solution (for reviews see for example [70–72]). With regards to proteins in the solid state, Venu et al. [73] investigated on hydration of crystalline BPTI using magnetic relaxation dispersion. Slow amide proton exchange in BPTI and lysozyme crystals was monitored by NMR [74,75]. Despite these pioneering studies, very little is still known about the dynamics of water interactions in immobilized protein samples. Recently, several groups reported the observation of water–protein interactions by site-resolved high resolution SSNMR [76,77]. Indeed, by recent progress in proton spectroscopy, these interactions can be probed looking at the water proton frequency in 2D heteronuclear correlation spectra, as commonly done in liquid state NMR. Fig. 6 shows a 2D 1H–13C correlation spectrum of Crh, with water–protein cross signals highlighted in gray.

2D 1H–13C HETCOR correlation spectrum. Cross signals between water protons and protein carbons can be observed at the ω1 water frequency, highlighted in gray (a). In b is shown the extract at the water frequency of the full carbon-13 spectral window [76]. (Reprinted with permission from J. Am. Chem. Soc. 126 (2003) 13336 Copyright (2003) American Chemical Society).
Site-specific assignments of these cross signals is in principle possible using 3D spectroscopy. Selective experiments are an interesting alternative as they are less time-consuming; selection of the water protons is possible in SSNMR by using their relaxation properties, e.g. their long T2 relaxation times compared to those of the protein protons [78]. Addition of a 13C–13C magnetization transfer step results in 2D spectra selective at the water frequency. It has been shown for the Crh protein that these spectra allow residue-specific assignments of the exchanging protons, and that the observed properties can be linked to structural features of the protein (Böckmann et al., J. Biomol. NMR, accepted). Indeed, cross signals could be observed between water and accessible threonine and serine hydroxyl protons, indicating fast exchange on the ms time scale; in contrast, hydroxyl protons involved in hydrogen bonds did not yield cross signals, indicating exchange on a slower time-scale.
Water–protein cross signals can also originate from two alternative magnetization transfer pathways. NOE interactions have been proposed to be at the origin of cross signals observed in ubiquitin [77], and dipolar water–protein interactions have been discussed for SH3 [79]. Further experimental evidence will be needed to settle on the origins of the water–protein cross signals observed in the different solid proteins.
11 Membrane systems
Membrane proteins are one of the ultimate targets of high-resolution SSNMR structural investigations. Studies of oriented membrane proteins and peptides have yielded the high-resolution structure of the gramicidin A channel in lipid bilayers [80] and the backbone structures of several single transmembrane helix peptides [81,82]. Rather than exploiting sample orientation, MAS experiments average orientation-dependent interactions as an alternative approach to achieve narrow resonances, thereby increasing resolution and sensitivity in the spectrum. Several groups are currently investigating selectively labeled membrane peptides and proteins by high-resolution MAS SSNMR [83–96], or investigate on general features of these proteins [97–101]. The ability of SSNMR to report on structural features of fully labeled ligands interacting with integral membrane proteins was demonstrated for several systems. The conformation of microgram quantities of neurotensin bound to its G protein-coupled receptor [35] has been analyzed, using double quantum spectroscopy to site-specifically assign the ligand. The dihedral angles predicted starting from the chemical shift information allowed to construct the backbone model of the ligand in complex with the receptor. Another examples are the 1H and 13C resonance assignments obtained on a uniformly 13C-labeled retinal chromophore, the covalently bound natural ligand of rhodopsin. Comparison to resonance assignments obtained on retinal in free form were qualitatively interpreted in terms of conformational rearrangements due to retinal–receptor interactions [102]. Neurotoxin bound to nicotinic acetylcholine receptor [103], as well as bacteriochlorophylls in light-harvesting complexes [21] are other examples. Partial site-specific assignments have been obtained for some uniformly enriched integral membrane proteins [104], indicating that structural characterization of such systems should be possible. However, many studies are still hampered the moment being by the difficulty to overproduce sufficient amounts of membrane proteins in bacteria.
12 Protein fibers
Beginning with the work of Lansbury et al. [105], who used SSNMR data to construct a model for amyloid fibrils formed by residues 34–42 of the full-length β-amyloid peptide (A(β)) associated with Alzheimer's, and Benzinger et al. [106,107], who first demonstrated the existence of cross-β structures with parallel β-sheets in their studies of amyloid fibrils, it has been found that amyloid fibrils in general are amenable to the most sophisticated SSNMR methods and that these methods yield structural constraints at a level of detail that has otherwise been inaccessible. An important body of work by Tycko and coworkers has led to a better structural understanding of the Aβ1–40 peptide [108–110]. The peptide conformation of amyloid fibrils formed by residues 105–115 of transthyretin was determined by Griffin and coworkers to high resolution using a combination of inter-atomic distance, torsion angle and chemical shift measurements. For a recent review on SSNMR on amyloid fibrils see reference [111].
13 Conclusion
Tremendous progress has been made in the last years in protein SSNMR. The advent of fast MAS, high decoupling powers and the development of multidimensional correlation techniques has lead to the possibility to investigate fully or extensively 15N/13C labeled proteins. This has opened the way to the detailed analysis of protein structure and dynamics by SSNMR. Resonance assignments have revealed a site-resolved picture of molecular interactions, involving proteins, ligands, and solvent. Very recent developments in NMR methodology, as 3D correlation spectroscopy, proton spectroscopy, techniques for resolution enhancement, as well as the use of several combinations of 2H/15N/13C isotope labeling schemes for proteins point to an even wider spectrum of interesting applications in the future. Latest hardware developments, including ultra fast MAS probes and ultra-high field magnets, promise ever more exciting possibilities.
SSNMR has demonstrated its capacity as a tool for structural biology, and is ready to proceed to applications to its main targets, membrane interacting proteins and non-native protein forms. Pioneering studies are already under way in several laboratories, and more are coming within reach.
Acknowledgements
The author is grateful to all coworkers and collaborators who contributed to the studies cited here. This work was funded in part by CNRS (PICS No. 2424) and the French research ministry (ACI ‘Biologie cellulaire moléculaire et structurale 2003’).