

1 Introduction
This article is a review of the results recently obtained in our group on increased resolution of 13C spectra of oriented lipid bilayers [1], and on complete 1H and 13C assignment of cholesterol in liposomes by HR-MAS [2–4], ultimately leading to a better quantification of cholesterol's hydrogen-bonding partners [4,5].
Solid-state NMR spectroscopy is a powerful tool to study membrane molecule structure and interactions because it allows the study of amorphous and partly mobile biological solids directly in the liquid-crystalline lipid bilayer. To the spectroscopist, perhaps the most important characteristic of the membrane's lipid bilayer is its high degree of local orientational order. In ordered systems the orientation dependent spin interactions dominate the NMR spectra and the relaxation behaviour of the spin system. Two complementary approaches have been developed: on one side, one can take advantage of the residual anisotropic interactions, which are due to the incomplete motional averaging and extract orientational information (static solid-state NMR on oriented samples) and on the other side, one can suppress the residual anisotropic interactions, leaving only the isotropic parts of the chemical shift and J-couplings (Magic angle spinning solid-state NMR).
Solid-state NMR spectroscopy of oriented membrane samples provides a robust method for examining the structure, dynamics and orientation of membrane peptides [6–10] and sterols [11,12] and has emerged in recent years as a powerful technique to characterise structural constraints of small molecules at their binding sites within membrane proteins [13,14]. Static spectra of uniaxially oriented samples are characterised by one resonance frequency for each labelled site. Since the observed resonance frequencies depend on the orientation of the molecular sites relative to the magnetic field direction, they provide the orientational constraints used in structure determination. For instance, the orientation and the structure of a peptide in the membrane can been determined by 15N labelling of the amides or 13C labelling of the carbonyls in the peptide backbone [15,16], the sterol order parameters can be extracted from quadrupole splittings of specifically deuterated positions [17]. However, 13C and 15N resolution generally available on mechanically oriented samples is close to 1–2 ppm [7,18,19] for lipids (and even ~10 ppm for peptides) and it would be highly desirable to increase it by at least one order of magnitude. A part of our study was to understand the different sources of carbon line broadening in cholesterol/dimyristoyl-phosphatidylcholine (DMPC) oriented samples and to determine how to improve the resolution in the 13C spectra of oriented bilayer systems [1].
Magic angle spinning (MAS) techniques can be used to remove the residual anisotropic interactions, leaving only the isotropic parts of the chemical shift and J-couplings. In addition, it is possible to combine the two approaches, first removing the anisotropic interactions by MAS and then selectively reintroducing, for example, the dipolar interaction between selected spins and measuring the dipolar couplings. In 13C MAS NMR studies, it is common to combine cross-polarisation (CP), or insensitive nucleus enhancement by polarisation transfer (INEPT), with high-power proton decoupling to obtain high-resolution 13C spectra where only isotropic chemical shifts remain. The resolution obtainable in fluid membrane systems approaches that obtained in solution studies. This is especially true for membrane lipids, where line widths of less than 0.1 ppm can often be obtained [20–22]. In purely lipidic bilayers in the fluid phase, various rapid motions occur (fast axial diffusion, wobbling, internal segmental motions). In particular, fast axial diffusion converts the homogeneous proton dipolar broadening into an inhomogeneous broadening, which can be effectively averaged out by MAS [22]. This leads to an increase in 1H and 13C T2s, and it has been shown that common liquid state NMR techniques such as selective 1J scalar coupling INEPT are efficient [23–25]. The 13C NMR line widths for sterols, peptides or proteins in membranes can be significantly larger, however, depending on the size and the dynamics of the molecules [26–29]. Cholesterol dynamics is well described by a fast axial diffusion (which provides axial symmetry to all anisotropic interactions), combined with a small amplitude wobbling (the molecular order parameter of the diffusion axis is equal to 0.94 for DMPC/cholesterol in a 7/3 molar ratio at 310 K) [11,30,31]. But, contrary to protons of lipids, no internal motion within the four rings of cholesterol contributes further to the averaging of anisotropic interactions. We compared various pulse sequences in order to obtain well resolved 1H–13C 2D correlation on the rigid part of cholesterol molecule, when it is inserted in a DMPC/cholesterol 7/3 lipid mixture, i.e. at a cholesterol molar ratio which is typically found in mammalian plasma membranes. In particular, we have compared dipolar (CP) and scalar coupling (INEPT) based transfers of coherences, and we have assessed the usefulness of homonuclear decoupling in the proton dimension [2]. We also compared the sensitivity of the liquid-like direct detection scheme (HETCOR) to the indirect detection scheme (ge-HSQC) of a 1H–13C 2D chemical shift correlation on the rigid part of a cholesterol molecule [3]. Finally, the complete and unambiguous 1H and 13C assignment of cholesterol in DMPC membranes has been performed on the basis of a combination of 1D MAS carbon NMR spectra with various polarisation transfer schemes and 2D MAS 1H–13C, ge-HSQC and dipolar HETCOR [4,5].
2 High-resolution 13C NMR spectra on oriented lipid bilayers
Bilayers mechanically oriented between glass plates provide an excellent model of natural membrane. We have recently determined cholesterol order parameters in DMPC–13C4-cholesterol (see Fig. 5A for cholesterol structure and atom numbering) oriented bilayers from the C–H dipolar couplings using 2D PELF correlating 13C chemical shifts and H–C dipolar coupling [32] identical to those previously obtained with deuterium NMR using specifically deuterated cholesterol [11]. However, in this experiment we systematically observed that cholesterol-C4 resonance had an apparent line width (1.5 ppm) much larger than expected from its transverse relaxation time (T2) from which we expect a refocused line width of 0.21 ppm.
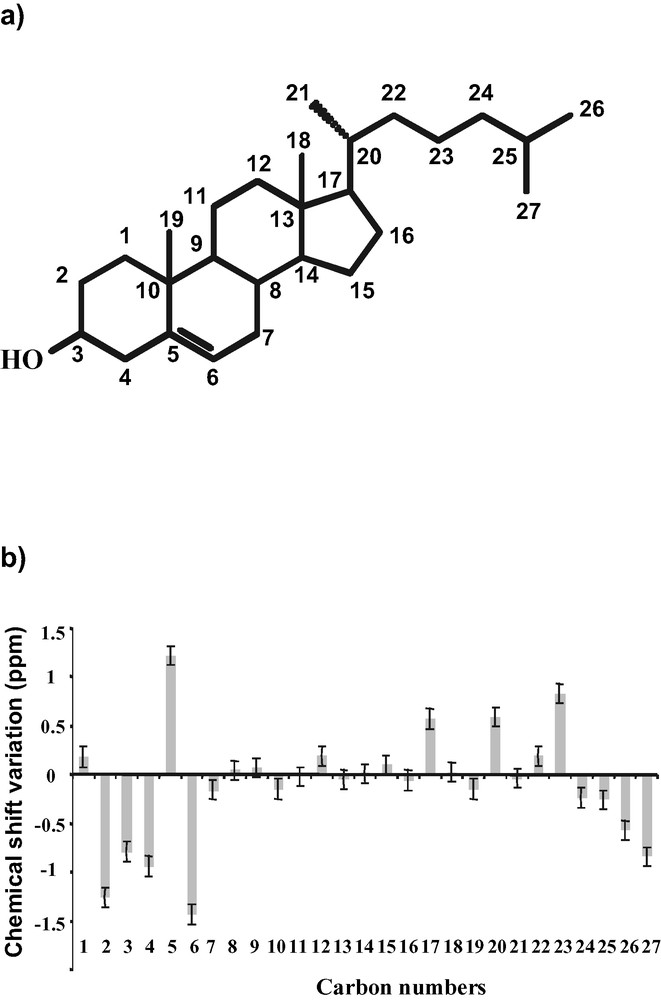
(a) Structure of cholesterol and classical carbon numbering. (b) Experimental chemical shift variation pattern between cholesterol in DMPC membrane and in CCl4 solution.
We performed a study on cholesterol/DMPC oriented sample in order to understand the different sources of carbon line broadening and to determine how to improve the resolution in the 13C spectra of oriented bilayer systems. We have specifically investigated the role of the sample geometry on B0 field inhomogeneities both theoretically and experimentally and the contribution of the quality of sample orientation (extent of liposomes and mosaic spread) [1]. We have prepared a DMPC [γ-13C]–13C4-cholesterol (7/3 mol/mol) sample, and oriented the lipid bilayers between glass plates so that the bilayer normal makes an angle of 90° (or of the magic angle) with B0. The magnetic field distribution within the sample, i.e. glass plates and multi-layers of lipids was calculated using the surface current formalism. It was interesting to observe that the overall field shape varies in a smooth manner over the whole sample. Therefore, we postulated that it should be possible to correct for induced field effects via careful shimming. The line shapes were simulated as a function of B0 field inhomogeneities and sample mosaic spread. Both effects contribute to the experimental line width. Using three signals of different CSA (13Cγ-choline, 13C4-cholesterol and 31P-DMPC), we have quantified both contributions and measured the mosaic spread accurately. It appeared that for bilayers oriented at the magic angle mosaic spread can play a significant role on line widths. At 90° and 0° orientations however, the major parameter is B0 field inhomogeneities and careful shimming on an internal signal is necessary to obtain the highest resolution. 13C-labelled choline methyl resonance of DMPC is a good candidate for this task. Fig. 1 shows the 13C CP solid-state NMR spectrum of 3/7 mol/mol cholesterol/DMPC-13Cγ-d54 oriented bilayers at 90° with respect to the external field. After optimisation of the important parameters (shimming on the choline resonance, mosaic spread of ± 0.30°), 13C line widths of 0.2–0.3 ppm have been obtained. The most intense signal is due to the choline methyl at 55 ppm (taken as an internal reference of chemical shift). Natural abundance phospholipid polar head group signals are also visible from 62 to 78 ppm. All other signals are due to the cholesterol carbon and C4 can be assigned to the resonance at 49.9 ppm according to the result obtained previously [1].
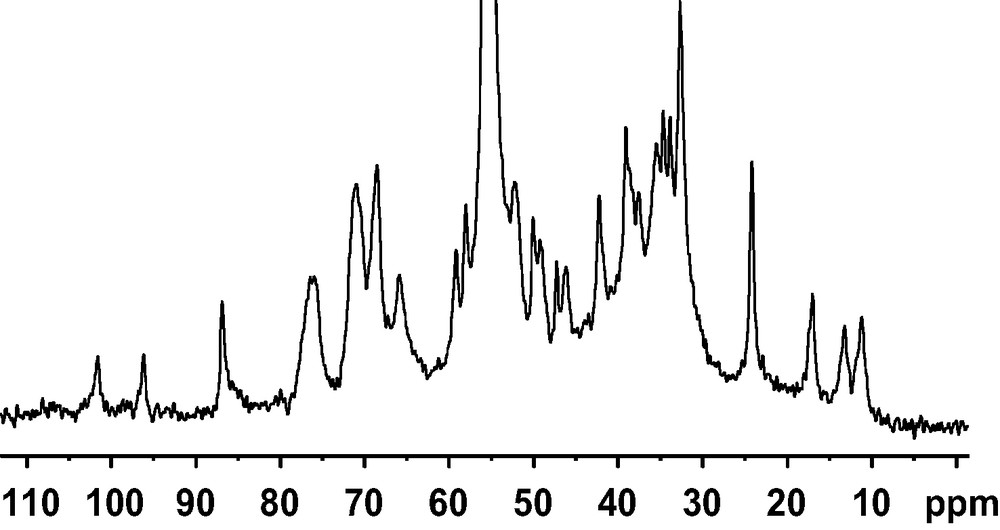
1D 13C CP spectrum on 30 mol% cholesterol/13Cγ-DMPC (2 mg cholesterol) oriented with the membrane normal perpendicular to the magnetic field, after shimming on the choline carbon resonance at 313 K.
13C spectrum was performed on a Bruker DMX narrow-bore spectrometer operating at 500 MHz for 1H, and recorded with a Bruker 7-mm double resonance probe with a solenoid coil oriented at 90° with respect to magnetic field. It was acquired with a standard cross polarisation experiment. A rf field of 55 kHz was used for cross polarisation (CP time 600 μs), whereas a proton rf field of 66 kHz was applied for decoupling during acquisition (20 ms, TD 4 K, DW 5 μs). The relaxation delay was 5 s and an exponential apodisation of 10 Hz was used prior to Fourier transformation. Carbon chemical shifts were referenced relative to the internal choline resonance taken at 55 ppm.
This study put forward that shimming directly on a sample resonance is required to get the highest resolution on oriented bilayer 13C spectra of lipids. It should be stressed at this point that we have been using a DMPC/cholesterol sample in the fluid phase which is known to orient particularly well, in agreement with the 0.3° mosaic spread found herein, and that the carbon resonances observed have sharp intrinsic line widths (< 0.2 ppm). The situation may be very different for peptide-membrane samples. First, it should be noted that all our analyses were performed in the assumption that the bilayer normal is a fast diffusion axis for all molecules, which is valid for cholesterol in DMPC, but certain peptides/proteins may behave differently (for example see [33,34]). The treatment of mosaic spread effect is very different in the rigid body hypothesis [35]. The typical T2-dominated line widths of peptides and proteins in membranes are often higher than 1 ppm [7,18,19]. Peptides/proteins mosaic spreads are often in a 5–20° range, which combined with a 15N CSA of about 170 ppm make it a dominant parameter. In some cases, lipids and peptides even display a different mosaic spread within the same sample [13,14,16,36,37], or peptides may display topological equilibria related to their function [38]. With all these restrictions in mind, it may nevertheless be useful to pay more attention to shims whenever the T2-dominated line widths are smaller than 1 ppm.
3 High-resolution 13C MAS NMR in model membrane: complete 1H and 13C assignments of cholesterol in lipid bilayer
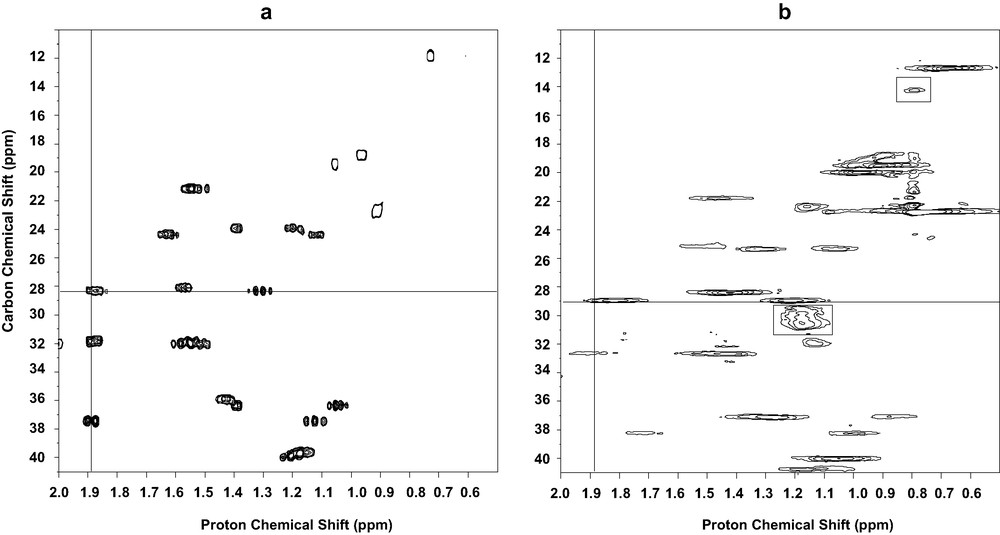
High-resolution 2D MAS NMR spectra of liposomes, in particular 1H–13C chemical shifts correlations have been obtained on fluid lipid bilayers made of pure phospholipids for several years. We have investigated the possibility to obtain high-resolution 2D MAS spectra of cholesterol embedded in membranes, i.e. on a rigid molecule whose dynamics is characterised mainly by axial diffusion without internal segmental mobility [2]. The sample was cholesterol-13C3,4 inserted at 30 mol% in DMPC liposomes. The efficiency of various pulse sequences for heteronuclear HETCOR has been compared in terms of resolution, sensitivity and selectivity, using either cross polarisation or INEPT for coherence transfer, and with or without MREV-8 homonuclear decoupling during T1. At moderately high spinning speed (9 kHz), a similar resolution is obtained in all cases (0.2 ppm for 1H3,4, 0.15 ppm for 13C3,4 cholesterol resonances), while sensitivity increases in the order: INEPT << CP (× 4) < CP + MREV. Again due to the specific dynamics of cholesterol on model membranes, it was shown that homonuclear decoupling during the proton evolution is not an absolute requirement and that when an MREV-8 sequence is employed it improves the effective resolution and sensitivity only marginally. Both cross polarisation and INEPT can be used for coherence transfer, the former making the experiment more sensitive by a factor 4, the latter allowing for a fully selective transfer between directly bound atoms. Since the four rings of cholesterol form a rigid part of the molecule and thus present the same dynamical properties [11], these conclusions, based mainly on the analyses of correlation peaks in position 3 (CH) and 4 (CH2), may safely extended to other positions in the rings. Since most of the H–H dipolar couplings in cholesterol are smaller than 10 kHz (Fig. 2), they are effectively averaged out by MAS at 9-kHz spinning speed and virtually no rotational side bands were observed along the proton dimension. This was not the case at 5-kHz spinning speed, and these side band patterns could be observed with a larger spectral width in the proton dimension. At this reduced spinning speed, the homonuclear dipolar coupling between the two geminal protons attached to C4 gives rise to spinning side bands from which one can estimate a H–H dipolar coupling of 10 kHz, which is in good agreement with the known dynamics of cholesterol in membranes.
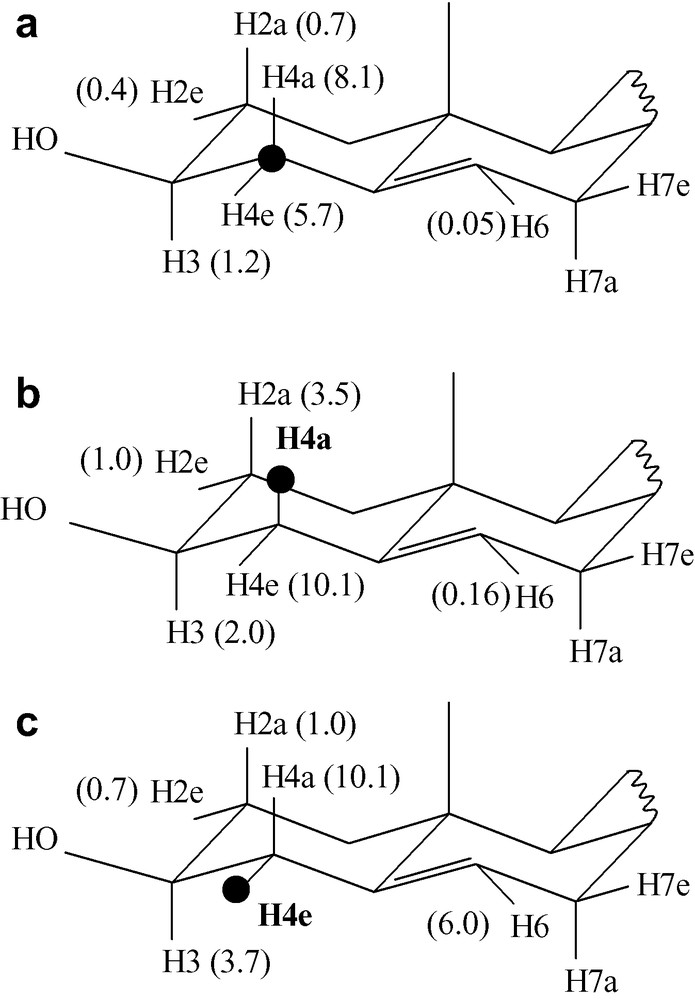
Calculated values of dipolar couplings within cholesterol in fluid bilayers (these couplings, in kHz, are those expected for bilayer normal oriented at 90° with respect to the magnetic field). (a) Heteronuclear dipolar coupling between carbon C4 and the closest protons, (b) homonuclear dipolar couplings between H4a and the closest protons, (c) homonuclear dipolar couplings between H4e and the closest protons. These couplings were calculated from the average orientation and dynamics of cholesterol in DMPC / cholesterol 7/3 mol/mol bilayers at 310 K, which have been previously determined accurately using deuterium NMR [11].
We have also shown that HSQC experiments of natural abundance cholesterol in liquid-crystalline DMPC membranes at 30 mol%, with a good resolution in both dimensions, can be obtained by HR-MAS experiments [3]. The inverse detection scheme is very suitable to enhance sensitivity on such samples and has allowed the detection of all the cholesterol expected cross peaks. Obviously, spinning rate is a key factor for an efficient transfer of coherence during the overall pulse sequence. The heteronuclear scalar composite pulse decoupling scheme GARP was found to be the most efficient technique for carbon decoupling during acquisition time. It can be foreseen that the gain in sensitivity brought by the use of a gradient HR-MAS probe (or a CPMAS probe with lock and gradients) and inverse detection in the HSQC experiment will play a key role in assignment efforts and analysis of fluid membrane components. A comparison between an HSQC of pure cholesterol typically obtained in organic solvent and the HSQC presently obtained, with cholesterol inserted in lipid membranes is represented in Fig. 3. Fig. 3B shows that, at 15-kHz spinning speed, several natural abundance cross peaks between directly bound carbon and protons can be observed with a good resolution and accuracy. Line widths obtained in the proton dimension were close to 0.1 ppm. In the carbon dimension, T2 relaxation time restricts the line width to 0.2 ppm. In this experiment, there was sufficient sensitivity to detect all the 24 expected natural abundance cross peaks, and the resolution was sufficient to resolve 20 out of them. Although a definitive assignment cannot be achieved with this experiment alone, detection of all heteronuclei and directly bound protons is usually the first step in this process. Since the relatively fast relaxation of coherences involved in the polarisation transfer would probably prevent the use of a liquid-like HMBC experiment, CP based HETCOR experiments with long contact time were required to establish connectivity between unbound atoms [4]. Fig. 4 illustrates the assignment of cholesterol from the combination of HSQC and dipolar HETCOR experiments. Using this ‘HMBC-like’ strategy, the complete proton and carbon assignment of cholesterol resonances in DMPC membrane was performed [4]. The variation between cholesterol carbon chemical shifts obtained in membrane and those obtained in anhydrous CCl4 is represented in Fig. 5B.
(a) Typical 2D 13C–1H HSQC spectrum of cholesterol in CDCl3 centred around the aliphatic region and referenced to TMS; Temperature: 310 K. (b) Same region of a 2D 13C–1H HSQC spectrum of cholesterol in DMPC-d54, at the same temperature and a MAS spinning rate of 15 kHz. Carbon spectral width 25 kHz. GARP carbon decoupling with a 13C π/2 pulse of 100 μs. A total of 1024 T1 increments with 64 scans each were collected. Square boxes surround cross peaks corresponding to the lipids. All the other cross peaks correspond to cholesterol resonances. Proton and carbon chemical shifts were internally referenced relative to the choline γ-CH3 resonance taken at 55 ppm (13C) and 3.18 ppm (1H) [2]. The line widths were equal to 0.1 ppm (CH, CH3) and 0.2 ppm (CH2) in the proton dimension and 0.2 ppm in the carbon dimension to be compared with 0.005 ppm for the corresponding liquid-state spectrum.
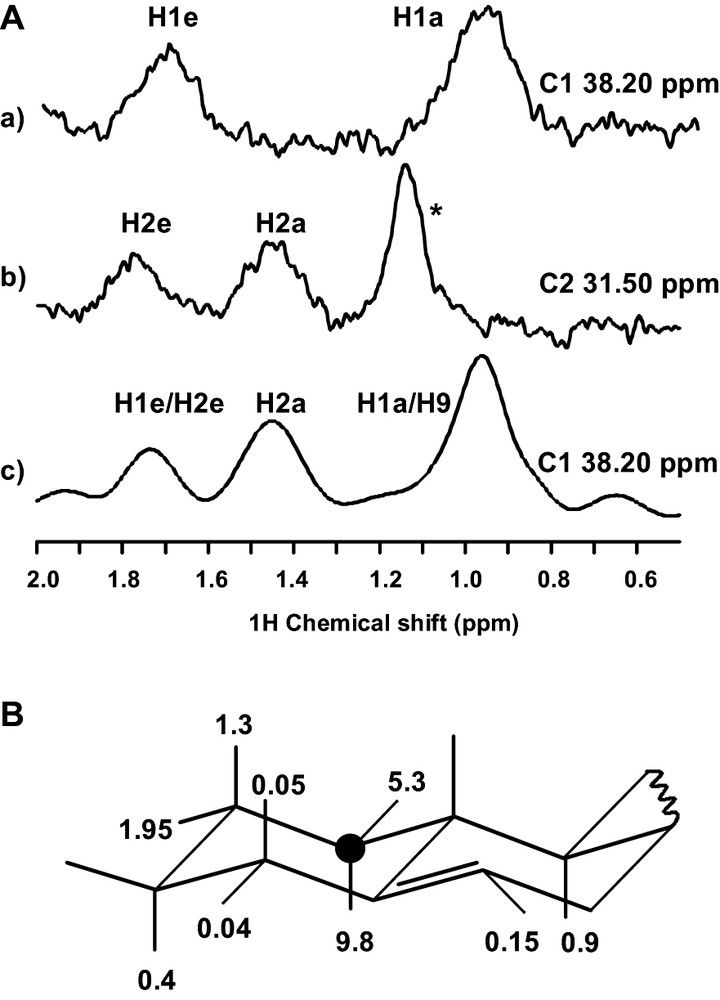
Combination of ge-HSQC and dipolar HETCOR for the assignment of cholesterol. (A) Cross-sections along the proton dimension at carbon chemical shifts corresponding to C1 (a, c) and C2 (b), extracted from ge-HSQC (cf. figure caption 3B for experimental conditions) (a, b) and Dipolar HETCOR (5-mm DOTY scientific XC-5 MAS probe, 9-kHz spinning rate, 2.5-ms cross polarisation contact time, 66-kHz 1H decoupling field and TPPM decoupling scheme during acquisition time) (c). Comparison of cross sections and favourable chemical shift dispersion allow assigning C1. * This signal arises from natural abundance lipid methylene. B Dipolar couplings between C1 and neighbouring protons (in kHz), calculated from the known structure and dynamics of cholesterol [11].
As shown in Fig. 4B the only protons displaying a significant (> 1 kHz) dipolar coupling with C1 are H1a,e and H2a,e. Correspondingly, the two HSQC traces in Figs. 4A reveal one-bond connections between C1 and the two H1 protons (a) and between C2 and the two H2 protons (b). The dipolar HETCOR trace along C1 contains connections with both H1 and H2 protons, thus guiding the assignment from C1 to H2 and C2 (c). It can be observed that, due to the 2.5-ms contact time, the signal intensities are not simply related the dipolar couplings in the 1–10-kHz range. Shorter mixing times (i.e. 100–500 μs) provide a more selective transfer, and give complementary information.
It has long been noticed that conformational effects (including dihedral angles, type of neighbouring atoms), hydrogen bonding or solvent effects influence the NMR chemical shifts. Therefore, it was interesting to analyse cholesterol's interaction with its environment on the basis of carbon chemical shift variation. The observed variations for carbons 17, 20–23 probably reflect a different conformation distribution of the side chain in membrane and in solution. Furthermore, magnitude and sign of the variations for carbons 1–6 can be interpreted in terms of C3–O3 rotameric distribution and in terms of hydrogen-bonding partners of the cholesterol hydroxyl group (these carbons are located in the first two rigid rings and no conformational effect is expected). We have first shown that the effects of H3 hydrogen-bonding and of C3-O3 rotamer distribution on chemical shifts could be accurately calculated by using a quantum-chemical approach [5]. Several hydrogen-bonding partners for cholesterol's hydroxyl group were considered, that is, water molecules (as hydrogen bond donor and acceptor), the acyl chain's ester bonds (modelled by acetone [5]) and non-ester phosphate oxygen atoms of the lipid polar headgroup (modelled by dimethyl phosphate [5]). The experimental chemical shift variations observed in membranes for carbons 1–6 were then analysed by combining the individual calculated chemical shifts variation for the three C3–O3 rotamer states and the four hydrogen-bonding partners [4]. We have thus directly demonstrated for the first time the interaction of the cholesterol H3 proton with the phosphatidylcholine phosphodiester, and quantified the percentage of this interaction in the membrane state (18 ± 3%, meaning that the OH group is bound to the phosphodiester 18% of the time). Other hydrogen-bonding partners have also been demonstrated, although their statistics have been quantified less accurately (Fig. 6). Our results show that less than one partner is in interaction with H3 (total H-bond via H3 = 61 ± 13%). This indicates that within our theoretical approach, lipids are not always H-bonded to cholesterol and that H3 can be free of any interaction, which may be related to the highly dynamic behaviour of cholesterol/DMPC system and its hydroxyl group. If one calculates the total number of H-bonded water, one obtains almost 1 water molecule per cholesterol, which is close to the value obtained by molecular dynamics simulations (1.1 ± 0.1) [39]. The total number of interactions (lipid esters, phosphatidylcholine phosphodiester, and water) obtained by our theoretical and statistical analysis is equal to 1.4 per cholesterol, which is again close to the 1.3 proposed by Pasenkiewicz-Gierula et al. [39]. Finally, we found in our data no convincing evidence that the rotamer distribution of the OH group is very different from what it is in solution, i.e. equal population of the three major rotamers.
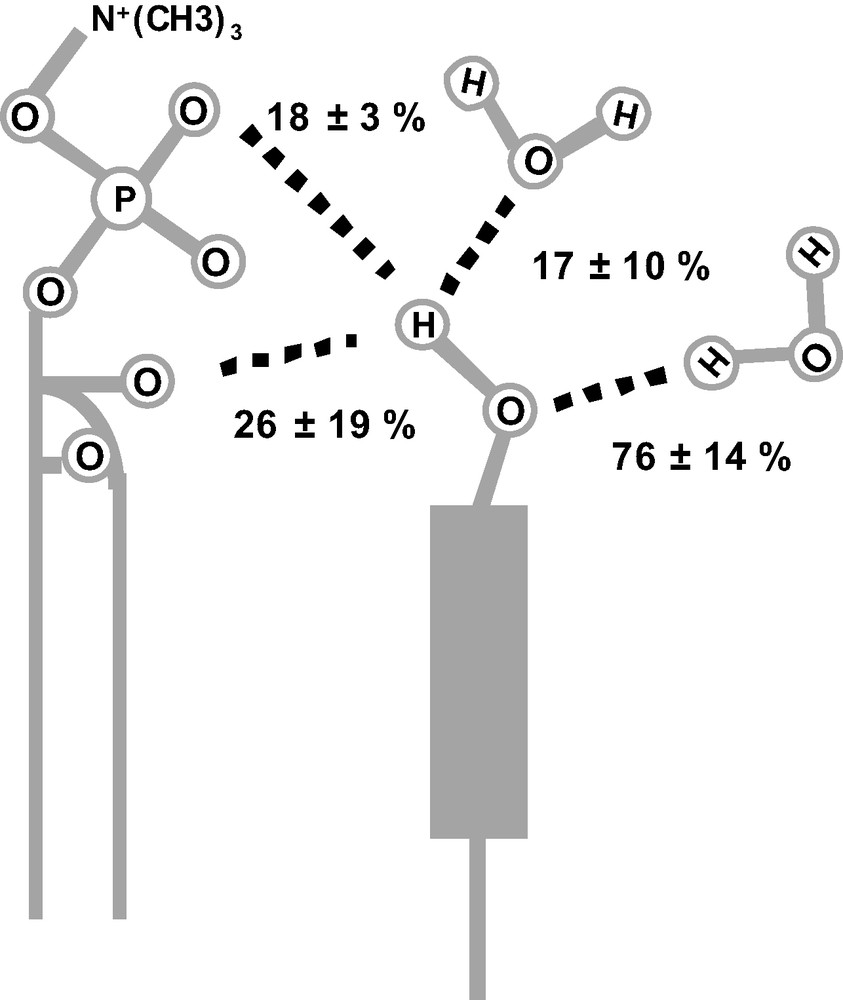
The number of hydrogen bonds per OH group was limited to one (or 100%) involving the hydrogen atom as a donor and two (or 200%) involving the oxygen atom as an acceptor (since the oxygen atom has two lone pairs).
Since the fitting procedure only imposed a maximum value of hydrogen-bonding partners, the final optimum result shows a certain percentage of OH groups free of hydrogen bonds. Note that the amount of hydrogen bonds with phosphate oxygen is particularly well defined.
We have thus established a new strategy, which allows to quantify the H-bonding behaviour of cholesterol in a membrane. This can now be applied to a series of interesting issues in membrane biophysics, such as the specific interaction of cholesterol with unsaturated lipids, with sphingomyelin in the context of raft formation, or the details of cholesterol's hydration as a function of the lipid mixture.