

1 Introduction
In plants, the oxidative dimerization of two phenylpropanoid units leads to a great variety of secondary metabolites. When the oxidative process implies the 8-8′ bonds, products are named lignans while the term neolignan is used to define all of the other type of linkage. Lignans and neolignans, found in a wide range of plant species, are responsible for their pharmacological activities and implied in their defense mechanisms [1,2]. Nevertheless, despite the importance of lignans in human health protection and in plant biology, systematic studies on the enzymes involved in their biosynthesis have only recently begun [3]. In this context it has been shown that phenylcoumaran benzylic ether reductases (PCBERs) that catalyze reductive process in 8-5′ linked lignans are ubiquitous vascular plant enzymes, which could have a role in plant defense. Flaxseed is known to be a rich source of lignans and Linum usitatissimum in vitro cultures have been established as a very useful tool to study the biosynthesis and the accumulation of metabolites [4–6].
In these cultures, a 8-5′ phenylpropanoid dimer (dehydrodiconiferyl alcohol-4-β-d-glucoside (DCG)) has been isolated and identified by nuclear magnetic resonance (NMR) spectroscopy. To evaluate the role of this compound in the cell cultures defense response, elicitation experiments by two fungal pathogens have been carried out. In these cultures DCG accumulation was quantified as a function of time and the gene expression of Lu PCBER was monitored by semi-quantitative reverse transcriptase polymerase chain reaction (RT-PCR).
2 Material and methods
2.1 Plant material
Cell suspension cultures of L. usitatissimum cv Barbara were established from hypocotyl-derived calli in MS-derived medium [7] supplemented with 3% sucrose, 8.88 μM benzylaminopurine (BAP) and 2.68 μM naphthaleneacetic acid (NAA). All suspension cultures were incubated on a gyratory shaker at 120 rpm in darkness at 25 °C and subcultured every 14 days.
2.2 DCG isolation and identification by NMR and mass spectrometry
A freeze-dried and ground sample of cells (5 g) cultured on MS-derived medium [7] was extracted with methanol/water (70/30, v/v) (200 ml) for 3 h at 60 °C. After filtration, the extract was concentrated to 15 ml. DCG was separated and purified by semi-preparative high pressure liquid chromatography (HPLC) using a μBondapack C18 prepacked column [10 μm, 25 × 100 mm], eluted with acetonitrile/H2O 0.2% acetic acid (15/85, v/v), flow rate 8 ml min−1, UV detection 280 nm, retention time 25 min. DCG was identified by 1H, 13C NMR and ESI-MS.
The 1 D and 2 D NMR spectra were recorded at 300 K on a Bruker Avance 300 spectrometer operating at 300.13 and 75.47 MHz, respectively, for 1H and 13C measurements. DMSO was used as the solvent and tetramethylsilane (TMS) as the internal standard. The chemical shifts are reported as parts per million (ppm) units relative to TMS. Mass spectral analysis was performed using a Finnigan SSQ 710 series mass spectrometer (LTE, UTC, France) in a solution of MeOH/H2O/CH3COOH (50:50:0.5 v/v).
2.3 Standard protocol for DCG quantitation
One gram, fresh weight (FW) of cells were separated from medium by filtration, rinsed with distilled water and freeze-dried. After extraction with methanol/water (70 /30, v/v) (10 ml) for 3 h at 60 °C and after filtration, the extracts were made up to 20 ml with distilled water and analyzed in triplicate by HPLC using a kromasil C18 column (5 μm, 250 × 4 mm, Macherey Nagel, France) eluted isocratically with acetonitrile/0.2% acetic acid/THF (20/79/1, v/v/v) at 280 nm with a flow rate of 0.7 ml min−1. In these conditions, the retention time was 15 min.
2.4 Elicitation of flax cell cultures
Fungal elicitors were prepared as described by Campbell and Ellis [8] with slight modifications. One of these fungal elicitors was obtained from mycelium extract of a specific pathogen of flax: Fusarium oxysporum F. sp. Lini, and the other from a mycelium extract of a broad-spectrum pathogen: Botrytis cinerea. Following transfer to fresh medium, flax cell cultures were inoculated at day 10 by fungal elicitors at a rate of 3% v/v. In the same way, controls were inoculated with sterile double distilled water and are used as reference during this study. Incubation continued on a gyratory shaker at 120 rpm in darkness at 25 °C. After treatment for a predetermined time, cells were collected for PCBER gene expression analyses.
2.5 Partial PCBER cDNA isolation and semi-quantitative reverse transcriptase-PCR
Total RNAs were purified from 150 mg of frozen tissues using the RNeasy Plant Kit (Qiagen). Degenerate PCR primers were designed to bind highly conserved sequences in plant PCBER genes. PCR product was resolved and purified from an agarose gel, subcloned by TA-cloning into pGEM-Teasy vector (Promega), sequenced and analyzed for sequences homologies by BLASTn and ClustalW programs.
For semi-quantitative RT-PCR analysis first-strand cDNA was synthesized from 1 μg of total RNA at 37 °C for 60 min, with 4 units of Omniscript Reverse Transcriptase (Qiagen). PCR was performed with specific primers designed from the partial cDNA sequence of Lu PCBER gene with PCBER-F sens primer (5′-TATGTCTCCGCAAACTGCTTTGC -3′) and PCBER-R antisens primer (5′-TTCAACAGTGGTGTATTTAACCTC-3′). cDNA fragments were amplified during 20, 25 and 30 cycles. To normalize the amount of mRNA in each PCR reaction, a PCR product (632 bp) corresponding to the Lu actin gene was amplified with ACT-F sens primer (5′-TCTGGAGATGGTGTGAGCCACAC-3′) and ACT-R antisens primer (5′-GGAAGGTACTGAGGGAGGCCAAG-3′). For this study 10 μl of 25-cycle PCR products were loaded on 1% (w:v) agarose gel separated by electrophoresis and stained with BET.
3 Results and discussion
In order to evaluate their content in lignans, the flax cell suspension cultures were analyzed by a HPLC method, as previously described [9]. The analysis revealed that these suspensions contained little amounts of lignans (data not shown) in regard to another compound accumulated at a very high level. This compound was isolated by semi-preparative HPLC and its structure elucidated on the basis of its NMR and MS spectral data.
3.1 NMR identification of a neolignan
The MS spectrum exhibited a molecular ion at m/z 538 [M + NH4]+ suggesting a molecular formula C26H32O11. The 1H NMR spectrum showed five aromatic protons: three at δ = 6.95 (d, J = 1.5 Hz), 7.06 (d, J = 8.0 Hz), and 6.84 (dd, J = 1.5, 8.0 Hz) and two at δ = 6.93 implying the presence of two aromatic rings, one with a 1,3,4-trisubstitution pattern and the second with a 1,3,4,5-tetrasubstitution pattern. Moreover, the spectrum showed signals due to two aromatic methoxyl groups at δ = 3.74 and 3.80 and two trans olefinic protons at δ = 6.21 and 6.45 with a pattern corresponding to an allylic alcohol. In addition, a glucose anomeric proton was observed at δ = 4.87.
The results of 1H and 13C NMR data and previous knowledge derived from lignans metabolites suggested that the isolated compound belonged to the neolignan class with a 8-5′ bond and a 7-4′ ether bond. The 13C NMR spectrum supported the presence of four oxygenated aromatic carbons at δ = 144.6, 147.8, 149.8, and 147.1. Complete assignment of the proton spin systems was achieved by DQF-COSY (Fig. 1a), while carbons were assigned from HSQC and HMBC spectra (Fig. 1b). The evaluation of the HMBC spectrum permitted the identification of the aromatic hydroxyl and O-methyl positions. In particular, the HMBC spectrum showed a weak correlation between the anomeric protons at δ = 4.87 and a quaternary carbon at δ = 147.1 indicating that the sugar was located at C-4. All the chemical shifts are given in Table 1. These data allowed, to identify this compound as the dehydrodiconiferyl alcohol-4-β-d-glucoside (DCG), a neolignan, which belongs to the phenylcoumaran family already described by Changzeng et al. [10]. In order to evaluate its involvement in plant defense mechanism, flax suspension cells were elicited with fungal pathogens (F. oxysporum F. sp. Lini, and B. cinerea).
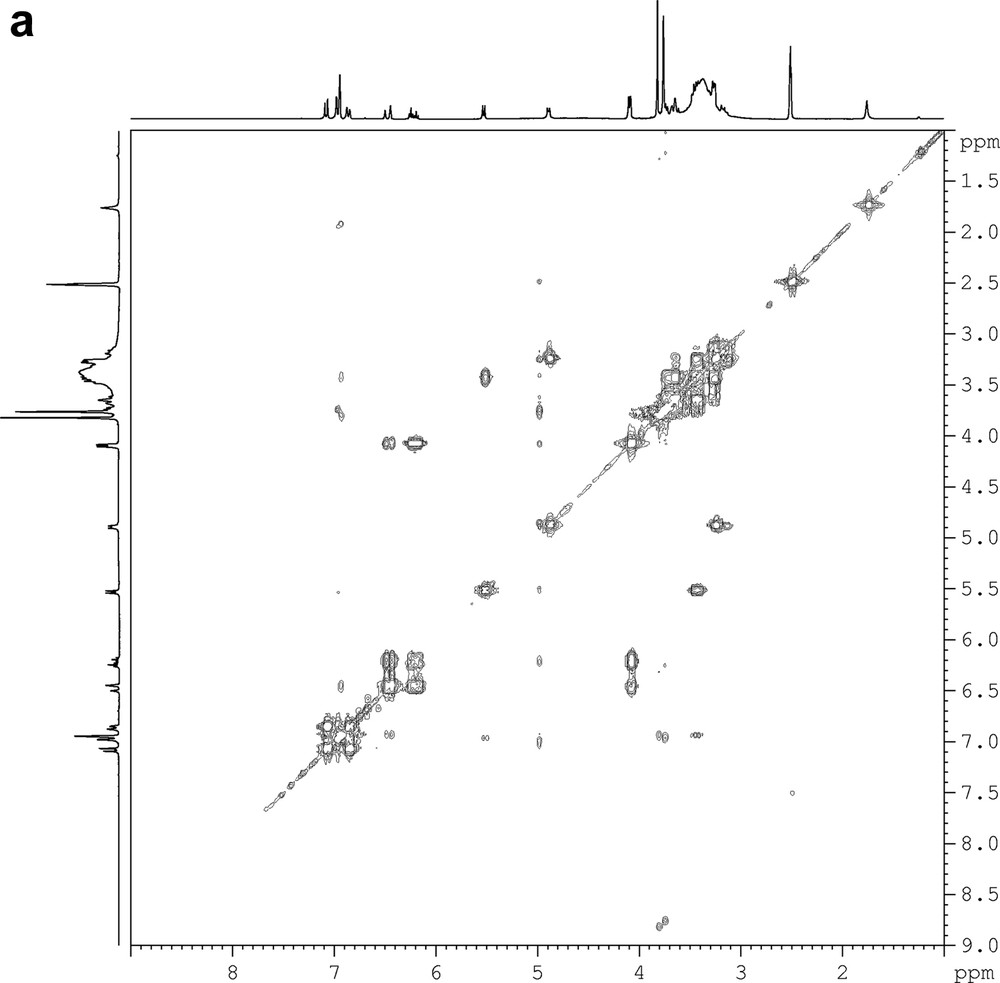
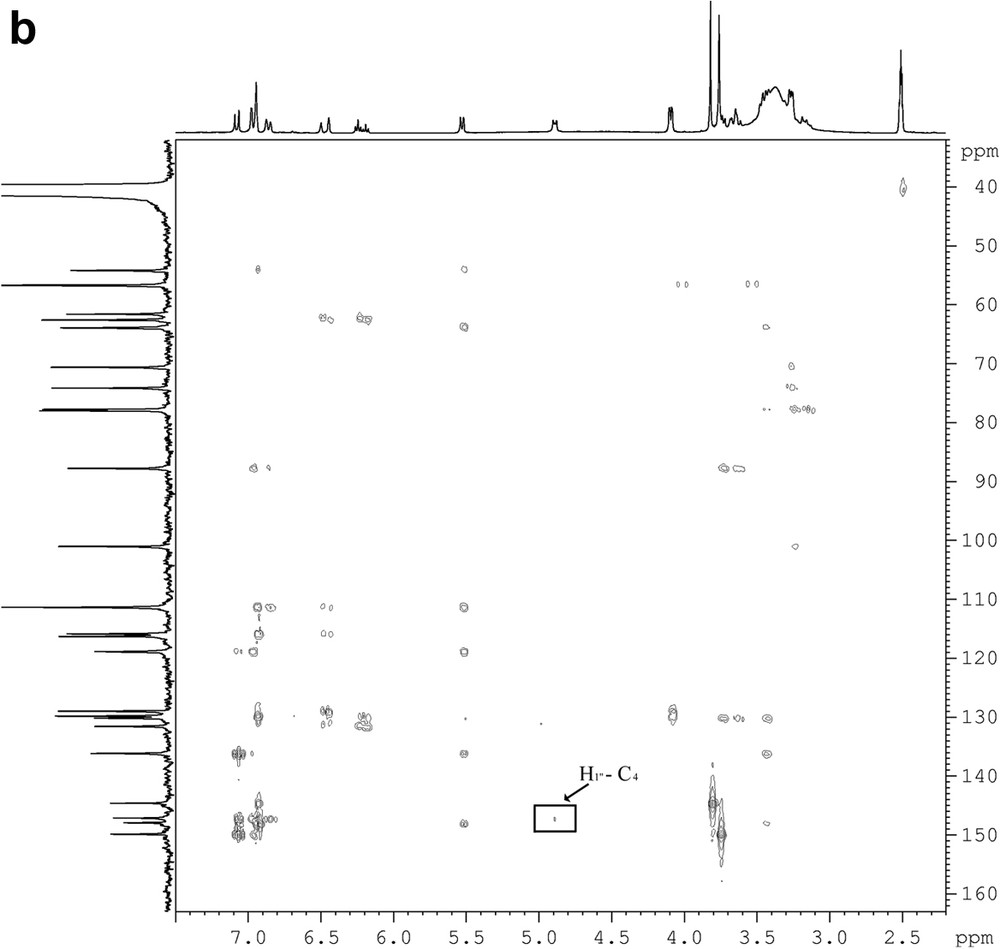
DQF-COSY (a) and HMBC (b) spectra of a pure fraction of dehydrodiconiferyl alcohol-4-β-d-glucoside (DCG).
NMR spectral data of dehydrodiconiferyl alcohol-4-β-d-glucoside (DCG)
| 1H | 13C | ||
| 1 | – | 136.2 | |
| 2 | 6.95 | 111.3 | |
| 3 | – | 149.8 | |
| 4 | – | 147.1 | |
| 5 | 7.06 | 116.2 | |
| 6 | 6.84 | 118.9 | |
| 7 | 5.51 | 87.7 | |
| 8 | 3.43 | 54.2 | |
| 9 | 3.63; 3.72 | 63.8 | |
| OCaH3 | 3.74 | 56.6 | |
| 1′ | – | 131.5 | |
| 2′ | 6.92 | 111.2 | |
| 3′ | – | 144.6 | |
| 4′ | – | 147.8 | |
| 5′ | – | 130.2 | |
| 6′ | 6.93 | 115.9 | |
| 7′ | 6.45 | 129.8 | |
| 8′ | 6.21 | 128.9 | |
| 9′ | 4.07 | 62.5 | |
| OCbH3 | 3.80 | 56.5 | |
| 1″ | 4.87 | 100.9 | |
| 2″ | 3.23 | 74.1 | |
| 3″ | 3.25 | 77.7 | |
| 4″ | 3.14 | 70.5 | |
| 5″ | 3.25 | 77.9 | |
| 6″ | 3.65; 3.43 | 61.5 |
3.2 Elicitation and RT-PCR semi-quantitative LuPCBER expression
After 96 h of treatment, the DCG content decreased by a factor of 13.1 and a factor of 2.1 when respectively treated with F. oxysporum F. sp. Lini and B. cinerea.
In order to monitor the effect of elicitation on phenylcoumaran lignans biosynthetic gene expression, degenerated primers were designed from global alignments of orthologos sequences of PCBER for PCR cloning. Indeed, PCBER catalyses the enantiospecific NADPH-dependent reduction of dehydrodiconiferyl alcohol and dihydrodehydrodiconiferyl alcohol (Fig. 2).

Biosynthetic pathway to the 8-5′-linked lignan of Pinus taeda showing the reductions steps catalyzed by PCBER as described by Kwon et al. [11].
Based on cDNA sequences of conserved regions of plant PCBER, degenerated primers were designed and synthesized for amplification of a putative partial LuPCBER cDNA fragment. A putative LuPCBER partial cDNA of 625 bp was obtained, sequenced and analyzed for homology studies by local alignments (BLASTn and ClustalW) and submitted to GenBank under the accession number AY837829. The deduced amino acid sequence was determined and database retrieval with PSI-BLAST as well as multiple alignments revealed that LuPCBER is highly similar to many other PCBER, isoflavone reductase (IFR) and Pinoresinol lariciresinol reductase (PLR) from higher plants at the amino acid level, indicating that the LuPCBER belongs to the IFR superfamily (Table 2).
BLASTp results showing identity and homology percentages of LuPCBER with other PCBER, IFR and PLR sequences (accession number from GenBank)
| Organism | Enzyme | Accession number | Identity (%) | Homology (%) |
| Tsuga heterophylla | PCBER | AAF64178 | 71 | 80 |
| Forsythia x intermedia | PCBER | AAF64174 | 70 | 83 |
| Glycine max | IFR | AAF17578 | 70 | 83 |
| Populus tricocarpa | PCBER | CAA06706 | 67 | 81 |
| Solanum tuberosum | PCBER | CAA63056 | 67 | 80 |
| Pinus taeda | PCBER | AAC32591 | 67 | 78 |
| Pinus pinaster | PLR | AL751330 | 67 | 78 |
| Forsythia x intermedia | PLR | AAC49608 | 49 | 62 |
| Thuja plicata | PLR | AAF63310 | 47 | 63 |
| Tsuga heterophylla | PLR | AAF64184 | 47 | 62 |
4 Conclusion
The LuPCBER transcript accumulation in response to elicitation with fungal extracts of B. cinerea and F. oxysporum was monitored by semi-quantitative RT-PCR (Fig. 3). It revealed a marked elicitor effect on the steady state mRNA levels corresponding to the LuPCBER gene. A rapid and continuous induction of LuPCBER gene expression from 1 h to 96 h post elicitation was observed in both elicited cell cultures, this was particularly marked for Fusarium treated cultures. Given the reaction catalyzed by PCBER, the decrease of dehydroconiferyl alcohol accumulated as glucosides in flax cell suspension could be correlated with the increase in LuPCBER gene expression and would therefore suggest the activation of PCBER in plant defense mechanisms [3].
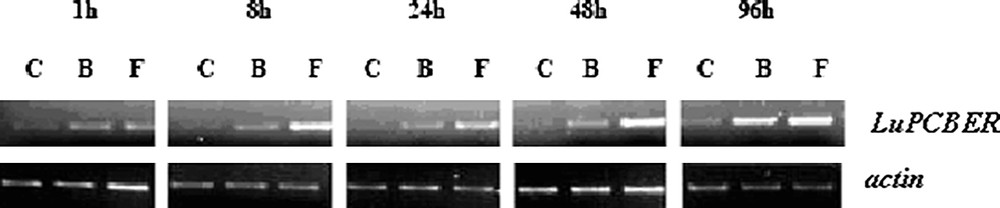
RT-PCR analysis of LuPCBER gene expression in control and elicited cell suspension cultures. Total RNA isolated 1, 8, 24, 48, and 96 h post elicitation was subjected to RT-PCR semi-quantitative analysis using an actin gene as internal control. C, control cell suspension, B and F, cell suspensions treated with mycelium extract of B. cinerea, or F. oxysporum.
Acknowledgements
J. Attoumbre wishes to thank the GERM association for a grant for his participation at the 19th annual meeting of the GERM.