

1 Introduction
Nuclear magnetic resonance (NMR) spectroscopy (in the biomedical field commonly referred to as magnetic resonance spectroscopy (MRS)) has been used for several decades to determine tissue metabolite levels in biological systems. These efforts were aimed for the most part at answering well-defined metabolic questions by analyzing specific biochemical pathways, e.g., energy metabolism, the Krebs cycle, or phospholipid (PL) synthesis and degradation (for an early review see Ross [1]). During the past years, more comprehensive metabolic MRS analyses of tissue metabolites were included. The term ‘metabolomics’ [2] is used for systematic and comprehensive studies of the metabolism of biological systems, including also statistical data evaluation [3]. Metabolomic MRS studies usually consist of simultaneous measurements of the concentrations of a large number of metabolites to determine metabolic profiles characteristic of biological systems.
MRS has been employed to study metabolic events characteristic of active, programmed cell death (apoptosis) vs. passive cell death (necrosis) (early review by Tozer et al. [4]). Apoptosis is an active process whereby a cell, upon receipt of an appropriate stimulus, engages a set of molecules that cooperate to dismantle the cell from within [5]. The purpose of monitoring metabolic NMR signals during apoptosis is twofold. First, it serves to elucidate the biochemical processes underlying apoptotic death. The knowledge gained from this research can then be exploited in the development of therapeutic treatment regimes targeting these metabolic processes. Second, MRS-detectable metabolic patterns characteristic of apoptosis may be used as markers for (i) diagnosis of disease, and/or (ii) treatment response, since numerous pathologies and therapies are associated with apoptotic events.
Most MRS studies of metabolic processes occurring during apoptosis have been carried out using cancer cells following treatment. Recent developments towards a global, metabolomic approach have shown that the systematic analysis of a large set of MRS-detectable metabolites may allow one to distinguish between cells that are sensitive to apoptosis induction and those that are resistant. The processes developing during apoptosis, or during resistance to apoptosis, can be monitored by metabolomic MRS as a function of time after treatment, in conjunction with measurements of complementary biological parameters.
2 Bioenergetic parameters in apoptosis
Studies of energy metabolism have been among the first reported metabolic MRS investigations in the context of apoptosis research [4]. These measurements are based on 31P NMR detection of well-known energy metabolites: adenosine di- and triphosphate (adenosine diphosphate (ADP) and adenosine triphosphate (ATP)), phosphocreatine (PCr), and inorganic phosphate (Pi). In fact, parameters indicative of the bioenergetic state can be directly calculated from these 31P signals. It should be noted that the nucleoside triphosphate (NTP) (NDP) peak detected by 31P MRS contains appreciable signals from guanylate nucleotides, guanylate triphosphate (GTP) guanylate diphosphate (GDP), and other NTP (NDP), and that their relative contributions to the observed peaks may vary, as was previously demonstrated by high-resolution 31P MRS of cell-extracts [4,6–9]. Fig. 1 depicts the fate of two common fuels, glucose and glutamine, in energy-producing pathways. The role of glucose in providing building blocks for purine and pyrimidine synthesis in nucleic acid production is also indicated in this figure.
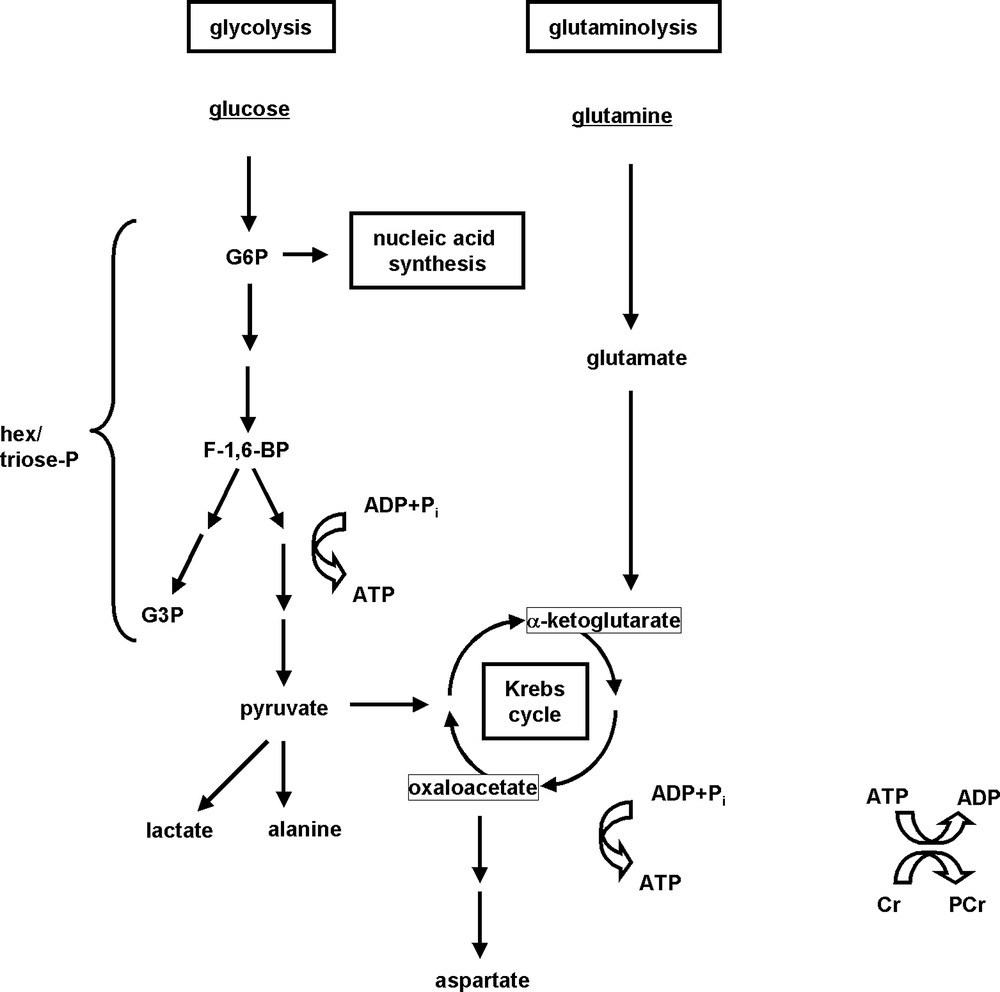
Simplified representation of glucose, glutamine and energy metabolisms.
Glucose and glutamine are highly utilized by mammalian cells as energy fuels, and produce high-energy ATP through mitochondrial oxidation (for glucose and glutamine), or through non-oxidative glycolysis resulting in lactate and/or alanine production (for glucose). Glucose also provides building blocks for purine and pyrimidine synthesis; these compounds serve to synthesize nucleic acids (mono-, di- and triphosphates of adenosine, guanosine, cytidine and uridine). PCr is another high-energy compound present in many cells. For abbreviations see list at the beginning of this article.
Post et al. reported decreased NTP levels in a dexamethasone-sensitive CEM variant after apoptosis-inducing treatment, but unchanged NTP levels for a dexamethasone-resistant CEM variant [10,11]. However, apoptosis induction in a different T-cell line (Jurkat cells transfected with ERtax, the tax oncogene of a human leukemia retrovirus) resulted in increased NTP levels in-vitro after apoptosis-inducing treatment with estrogen, after a temporary decrease [12]. A time course study has been performed by Viola et al. for an erythroleukemic cell line, TF1 (in-vitro 31P MRS of cell suspensions), serving as a model for apoptosis induction by photodynamic therapy (PDT) [13]. PDT over 1 h caused significant TF1 apoptosis and resulted in an increased NTP/Pi ratio. All three studies mentioned above were performed in-vitro on intact cells. A cell-extract study by Lutz et al., focusing on metabolic changes in HT-29 human colorectal carcinoma cells following apoptosis induction by cytokines (TNF-α/interferon-γ (IFN-γ)), found progressively increasing NTP levels starting from 4 h out to 15 h of combination treatment, while no consistent trend was observed for PCr or Pi [14].
Treatment of a different human colorectal carcinoma cell line, SW620, with mechlorethamine (HN2) resulted in late-stage apoptotic cells that possessed intact membranes but were devoid of NTP (cell-extract study [15]). HN2 was also used to induce apoptosis in HT-29 cells [16]. In this work, an increase in NTP and PCr over time was reported for cell-extracts. Williams et al. reported some ATP reduction but no change in energy charge after apoptosis induction in the human promyelocytic leukemia cell line, HL-60, and the Chinese hamster ovary cell line, CHO-K1, by a variety of drugs (cell-extract study [17]). These examples clearly illustrate that apoptosis effects on energy metabolism vary as a function of the cell type/variant used, but also depend on the mode of apoptosis induction as well as on the time point chosen for quantitation of energy metabolites. Neither increases nor decreases in bioenergetic parameters per se can be considered characteristic features of programmed cell death.
3 Glucose metabolism in apoptosis
Energy metabolism is closely linked to glucose metabolism since glucose is one of the most important cellular energy sources. Consequently, several of the studies mentioned above included measurements of glucose metabolite levels. A comparison between human leukemic cells sensitive to apoptosis induction by dexamethasone and a resistant variant suggested that differences in glucose metabolism did not contribute to steroid resistance (13C NMR analysis of a suspensions of cells that had been incubated with [13C2]-labeled glucose) [18]. However, this report did not contain data on the evolution of glucose metabolism following treatment. Viola et al.'s in-vitro investigation of PDT of intact TF1 cells demonstrated that apoptosis induction did not affect UDP–hexose concentrations [13]. Nunn et al. reported a drop in lactate, the end product of non-oxidative glycolysis, in apoptotic vs. non-apoptotic neutrophils (1H MRS of cell-extracts [19,20]). In contrast, a roughly linear lactate increase over time (+50% after 15 h) was detected in-vitro by Lutz et al. [14] in HT-29 cells following apoptosis induction with TNF-α/IFN-γ, paralleled by a weaker NTP increase (combined 1H and 31P MRS of cell-extracts, see also previous section).
Two groups observed temporary but drastic increases in fructose 1,6-biphosphate (F-1,6-BP) signals in leukemia cells undergoing apoptosis (HL-60 cells treated in-vitro with etoposide or camptothecin, and L1210 cells treated with HN2; [17] and [15], respectively; 31P MRS of cell-extracts). However, for TNF-α/IFN-γ -treated HT-29 cells investigated by Lutz et al. [14], modest but sustained increases in F-1,6-BP and glycerol 3-phosphate (G3P) have been observed in early apoptosis, paralleled by increased NTP (cell-extracts).
4 Lipid metabolism in apoptosis
4.1 Phospholipid metabolism
A decrease of phospho(ryl)ethanolamine (PE) and phospho(ryl)choline (PC) has been demonstrated to precede cell death in several types of human cancerous cells in-vivo [21]. Subsequently, several groups investigated the hypothesis that a reduction in PC or phosphomonoester (PME) is a characteristic feature of cells undergoing apoptosis. Adebodun's early in-vitro study on intact cells found this to be the case for PME in apoptotic dexamethasone-treated human leukemic CEM cells; however, one of three steroid-resistant CEM variants also exhibited a PME decrease [11]. In Chinese hamster ovary cells, CHO-K1, and human promyelocytic leukemia cells, HL-60, a rather consistent decrease in PC but not in PE was observed in-vitro after apoptosis induction (cell-extracts [17]). The role of MRS-observable compounds of phospholipid metabolism is illustrated in Fig. 2 for the two most abundant PL, phosphatidylcholine and phosphatidylethanolamine.
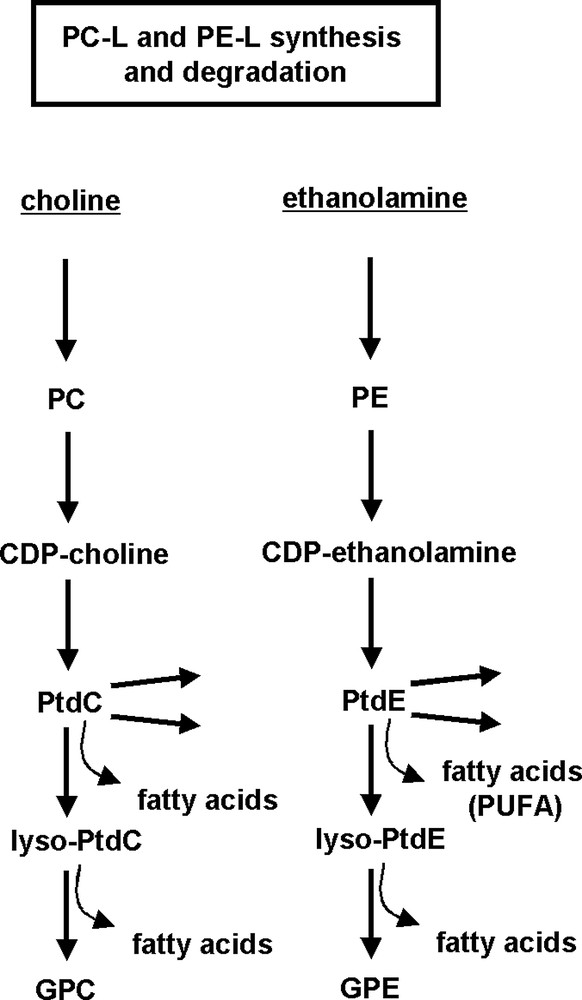
Simplified representation of phospholipid metabolism.
PL synthesis: Kennedy pathway is shown for choline and ethanolamine glycerophospholipids. PL degradation: fatty acids released through phospholipase A and lysophospholipase activity form triglycerides accumulating in form of intracellular lipid droplets. The latter can be detected by MRS of intact tissue. For abbreviations see list at the beginning of this article.
Viola et al. who investigated metabolic effects of relatively late-stage apoptosis in suspensions of intact erythroleukemic TF-1 cells, did not detect any significant PC or PE changes in PDT-treated apoptotic cells vs. non-apoptotic cells (in-vitro study [13]). Hakumaki et al. found a temporary PC and PE decrease at a rather advanced stage of apoptosis in BT4C glioma treated in-vivo with ganciclovir [22]. The time course study conducted by Lutz et al. over a 15-h period using colorectal HT-29 cells showed a consistent decrease in PC following apoptosis-inducing treatment with TNF-α/IFN-γ (cell-extracts [14]). These data were obtained from cells at a rather early stage of apoptosis. However, it should be noted that non-apoptotic HT-29 cells treated with TNF-α only also showed a PC drop, albeit to a lesser extent. The glycerophosphocholine (GPC)/PC ratio, which is frequently used as an indicator of changes in choline homeostasis, was transiently increased in apoptotic HT-29 cells treated with TNF-α/IFN-γ [14], and in miltefosine-treated apoptotic C6 glioma cells [23].
Spectacular CDP-choline increases were observed for CHO-K1 cells and, to a lesser extent, HL-60 cells in cell-extracts after apoptosis induction with several different drugs, and was considered to be indicative of an inhibition of phosphatidylcholine synthesis [17]. Viola et al. observed that in PDT-treated TF1 cells, apoptosis was accompanied by considerable CDP-choline accumulation (cell suspension study [13]). On the other hand, there are several examples of cell suspension and cell-extract 31P MRS studies of cells undergoing apoptosis without CDP-choline accumulation [11,14–16,24]. Thus, the possibility that PC or PC/GPC per se may be useful to distinguish cell death induction from non-lethal growth inhibition can be virtually excluded [25].
4.2 Mobile lipids (ML)
The first observation of a correlation between ML accumulation and apoptotic cell death has been published by Blankenberg et al. [26] for several leukemic cell lines and lymphoblasts after serum deprivation and glucocorticoid or doxorubicin treatment. An increase in lipid methyl (ca. 0.9 ppm) and, to a larger extent, aliphatic methylene (–(CH2)n–, ca. 1.2 ppm) signal was observed in in-vitro 1H NMR spectra of intact cells. The authors hypothesized that these increases, in particular an increasing aliph. CH2/CH3 ratio, may be characteristic of apoptotic vs. necrotic cell death. Subsequently, numerous reports confirmed that various forms of stress, even necrotic cell death, could induce intracellular accumulation of lipid droplets providing signals from mobile CH3 and aliphatic CH2 moieties [27].
Engelmann et al. found an increased biosynthesis of triacylglycerol and diacylglycerol in apoptotic KB cells, whereby apoptosis was induced by miltefosine [23]. However, these results were obtained on the basis of total-lipid extracts, which represent the entire cellular lipid pool, not only droplet-derived lipids.
Hakumaki et al. suggested that 1H MRS peaks specific of polyunsaturated fatty acids (PUFA) are specifically associated with apoptosis [28]. However, it should be noted that evidence for the hypothesis of an apoptosis-specific PUFA increase in lipid droplets is largely based on experiments with in-vivo or ex-vivo tumor tissue rather than cultured cells, thus complicating the identification of the precise origin of the detected PUFA signals [22,28,29].
As pointed out in the preceding sections, changes in individual MRS signals and metabolite concentrations are, in all likelihood, not characteristic traits of the metabolism of apoptosis [25]. Notwithstanding, a more comprehensive approach to MRS of apoptotic cells has the potential of revealing metabolic patterns leading to a deeper understanding of the cellular metabolism underlying programmed cell death. The following section will present recent studies approaching this goal.
5 Developments towards metabolomic MRS of apoptosis
In recent years MRS studies have begun to adopt an increasingly comprehensive approach to the analysis the metabolism of apoptotic cells. A number of methodoligical perequisites has been described elsewhere [30]. Multinuclear MRS has been used to quantitate a large number of metabolite peaks for different times following apoptosis induction, and the results obtained were subjected to systematic statistical evaluation [8,9,24,31–33]. For instance, one of these projects involves metabolite changes in BT4C rat glioma undergoing ganciclovir-thymidine kinase gene therapy-induced programmed cell death (1H and 31P MRS) [32,33]. This work combines in-vitro MRS of excised tissue samples (high-resolution MAS) and MRS of tissue extracts with in-vivo MRS. The authors found that resonances from mobile monounsaturated fatty acids and PUFA were best correlated with late apoptosis as detected by the TUNEL assay, whereas signals from water-soluble metabolites were not correlated with cell death induced by the gene therapy treatment chosen [32,33]. However, since solid tumor tissue rather than cultured cells were employed, some questions remain to be answered with respect to the origin of the observed signals, i.e. to what extent they actually represent glioma cells exposed to treatment, rather than macrophages, stroma and extracellular matrix components [34].
While the metabolomic MRS approach has been developed and extensively used by J. Nicholson's group [33] and others, it's application to apoptosis research has been rather limited thus far. For instance, the metabolism of an apoptosis-sensitive cancerous thymocyte culture (WEHI7.2) was compared with that of apoptosis-resistant variants (study by Lutz et al. [8]), and comprehensive time course studies were undertaken to follow metabolic changes upon dexamethasone treatment [9,24,31]. These metabolomic experiments were performed by using high-resolution 31P and 1H MRS of cell extracts, and were accompanied by measurements of enzyme activities, specific enzyme protein levels (immunoblots) and intracellular substrate uptake, but also by tests of mitochondrial function, cell cycle analysis and characterization of relatively early apoptosis stages by annexin-V labeling (flow cytometry) and cytochrome c release from the mitochondria into the cytosol. Results were evaluated by employing multiple-comparison and correlation statistics.
In these WEHI7.2 studies, lyso-phosphatidylcholine (lysoPtdC) levels turned out to be the metabolite that was best correlated with the percentage of apoptotic cells (r = 0.92) [24]. In addition, the increases observed in lysoPtdC concentration after steroid treatment were correlated with the steroid sensitivity of the WEHI7.2 variants such that the increase was seen earliest in the most steroid-sensitive WEHI7.2 cells, was delayed in a moderately resistant variant and showed no consistent change in an entirely steroid-resistant variant after dexamethasone treatment. Statistical significance of observed changes in lysoPtdC concentration vs. controls was confirmed by an analysis of variance (ANOVA) method. Due to the use of optimized sample preparation as well as spectrum acquisition and data processing methods, high reproducibility was achieved (S.D. < 0.1% of total PL) despite low lysoPtdC concentrations (between ca. 1 and 3 nmol/mg protein, corresponding to 0.5–1.6% of total PL) [24]. The analysis of several different steroid-resistant variants virtually excludes the possibility that non-toxic side effects of steroid treatment may produce a similar lysoPtdC accumulation in the cancerous thymocytes used. However, it remains to be determined if other cell lines and treatment regimes would produce a similar lysoPtdC effect as cells undergo apoptosis.
In addition to pairwise comparison between metabolite levels of treated and untreated cells for equal times of exposure to drugs, and the analysis of the time course of these differences for sensitive and resistant variants, also the examination of the metabolic drift over time in untreated cells was of interest. The ratio of choline-containing vs. ethanolamine-containing PL, PC-L/PE-L, decreased over time after medium replenishment in batch culture, and this trend increased with increasing resistance to apoptosis [24], confirming a previously described tendency towards lower PC-L/PE-L values for a broader range of WEHI7.2 cells resistant to oxidative stress [8]. A similar pattern of metabolic adaptation to progressively changing culture conditions has been observed for glutamine, aspartate, lactate and UDP–hexose concentrations as well, suggesting flexible adjustments of glutamine and glucose metabolisms in apoptosis-resistant cells [9]. Thus, the comprehensive metabolomic approach used in this study has revealed several global metabolic patterns hinting at a generally reduced metabolic flexibility in apoptosis-sensitive vs. resistant cells.
Time-course MRS studies of metabolite concentrations revealed that the resistant variants undergo increased glutaminolysis particularly after a long time in culture, as glucose levels in the culture medium decreased. The increased role of mitochondrial vs. non-oxidative ATP production, and of mitochondrial vs. cytosolic hexokinase, in apoptosis-resistant vs. sensitive cells was confirmed by determinations of mitochondrial and cytosolic hexokinase activities and protein expression [9,31].
Future metabolomic projects regarding apoptotic cells should:
- • systematically include multinuclear MRS to broaden the range of detected metabolites;
- • employ optimized sample preparation, as well as spectrum acquisition and processing techniques for highest spectral resolution, in order to optimize metabolite quantification (for example, spectra incl. detailed experimental protocols see [6,8,9]);
- • include the use of labeled metabolic substrates to determine their uptake into cells, and their conversion into specific metabolites (flux measurements);
- • systematically include time course MRS covering the relevant stages of the apoptotic process;
- • ensure high reproducibility of results by taking into account the metabolic drift occurring in batch cultures;
- • use appropriate apoptosis-resistant cells (e.g., variants) along with apoptosis-sensitive cells to distinguish between apoptosis-related treatment effects and apoptosis-independent side effects (potentially resistance-related);
- • further develop techniques for absolute metabolite quantitation (for high-resolution magic-angle spinning (HRMAS) of excised tissue samples and in-vivo MRS), and tools for statistical processing of ever growing multiparametric data sets;
- • and combine metabolite quantitation with enzyme studies and cell biological measurements such as the characterization of early and late apoptotic stages, to facilitate mechanistic interpretation of observed metabolic changes. The systematic integration of metabolomic MRS and other biochemical and biological methods will generate the synergies necessary to obtain fundamentally new insight into global metabolic patterns and mechanism(s) of programmed cell death.