

1 Introduction
The unique transcript produced from the integrated proviral cDNA of human immunodeficiency virus (HIV) serves as the genome for newly synthesized virions and also for production of mRNAs by alternative splicing [1]. Thus, retrovirus multiplication depends upon equilibrium between spliced and unspliced primary transcripts. Alternative pre mRNA splicing is achieved by the spliceosome, a macromolecular complex. It plays a central role in genetic regulation and is itself highly regulated. This regulation involves splice sites consensus sequences, RNA secondary structure that affect splice site recognition and RNA elements regulatory sequence that strongly affect spliceosome assembly called splicing enhancer or splicing silencer [2].
To date many exonic splicing enhancer (ESE) and exonic splicing silencer (ESS) have been identified HIV-1 RNA.
As part of a structural study of alternative splicing in HIV type 1 (HIV-1), we focused on one ESS sequence, the proximal exon splicing site two (pESS2). It is located within tat exon 2 and regulates the utilization of the A3 3'splicing site. It is part of a stem-loop structure (SLS2) that may influence its activity [3]. The nuclear component that binds pESS2 is heterogeneous ribonucleoprotein H (hnRNP H) [4,5]. This protein exhibits a modular structure with three RNA recognition motif (RRM) and low complexity glycine rich domains.
The biomolecules design strategy chosen to make the analysis feasible by NMR and their production is presented. The first NMR data establishing the RNA secondary structure is shown, along with the preliminary NMR results obtained with the protein constructs that found the feasibility of the study.
2 Materials and methods
2.1 NMR samples
2.1.1 Protein expression and purification
Overproduction of a protein corresponding to the first two RNA binding domains (RRM1,2) of the human hnRNP H and of each of these domains (RRM1 and RRM2) was achieved using a T7 expression system. The expression vector is a pET15b plasmid containing each construct chosen after bioinformatics analysis [6] of hnRNP H. BL21λDE3 Escherichia coli strains were transformed with the corresponding plasmid, including a N-terminal Histidine-Tag to facilitate purification.
E. coli cells were first grown on LB plates supplemented with 100 μg/ml ampicillin. A single colony was transferred to 20 ml LB medium containing 100 μg/ml ampicillin. This culture was grown to a density (estimated from A600 nm, the absorbance at 600 nm) of = 0.6 then transferred to 1 l of minimal medium containing 15NH4Cl and uniformly 13C labeled glucose in a 2-lfermbar. Cells were grown at 310 K with shaking (160 rpm) to a density of A600 nm = 0.7. Protein expression was then induced with 1 mM IPTG during 4 h. Cells were harvested by centrifugation at 277 K for 10 min at 6000 rpm. The cell pelled were conserved at 193 K before lysis.
The cells were resuspended in 80 ml cold buffer A (50 mM phosphate buffer pH = 8, 10 mM imidazole, 300 mMNaCl) and sonicated. This suspension was centrifuged for 20 min at 10 000 rpm. After sonication, the protein solution was loaded onto a Ni-NTA column at a flow rate of 1 ml/min. Weakly bound proteins were washed from the resin with buffer A. Increasing the imidazole concentration from 10 mM to 300 mM eluted the recombinant histidine-tagged protein. Proteins were then concentrated using Amicon Cell and Centriprep YM-3 and the buffer changed to a 10 mM sodium phosphate buffer (pH = 4).
2.1.2 RNA preparation and purification
RNA samples of the constructs derived from the wild type HIV-1 RNA stem-loop structure containing the A3 splicing site (Fig. 1) were prepared via in vitro transcription using T7 RNA polymerase and synthetic DNA templates using published procedures [8]. Samples were prepared unlabeled or uniformly 15N-labeled. Mutant RNA STEP2 without the bulge region and the smaller RNA STEP 1 were chemically synthesized on a Gene Assembler Plus (Pharmacia) using phosphoramidite chemistry. These RNA were only prepared unlabeled.
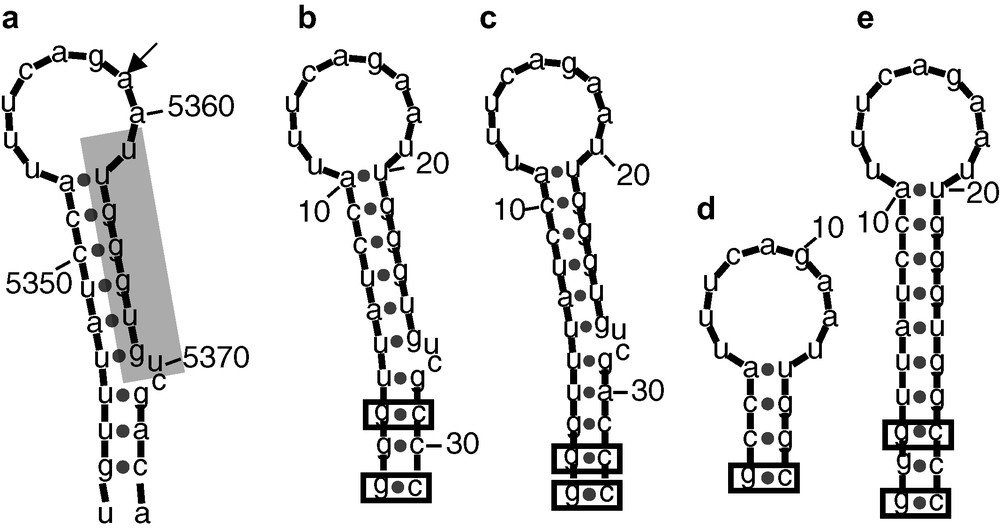
Mfold [7] predicted structures of the RNAs derived from the A3 splicing site. (a) Original HIV Bru sequence [3], the A3 splicing site is indicated by an arrow. The proximal exonic splicing site 2, pESS2 [5] is shaded. (b–c) A3_1 and A3_2 stem loop models engineered for the NMR studies. The base-pairs added to stabilize the structure are boxed. (d–e) STEP1 and STEP2 models used to facilitate the NMR signals assignment. STEP1 corresponds to the apical stemloop. The bulge has been deleted in STEP2. Masquer
Mfold [7] predicted structures of the RNAs derived from the A3 splicing site. (a) Original HIV Bru sequence [3], the A3 splicing site is indicated by an arrow. The proximal exonic splicing site 2, pESS2 [5] is ... Lire la suite
Only enzymatically produced RNA were purified using 20% urea polyacrylamide gel and electroelution.
2.2 NMR experiments
For the NMR experiments in H2O, the RNAs were dried and re-dissolved in 5% D2O/95% H2O. RNA sample concentrations were kept at 0.5 mM to limit duplex formation. The pH of all the solutions was set to 6.5 with a 10 mM sodium phosphate buffer. 1H–1H and 1H–15N NMR spectra were recorded on a Bruker DRX-600 with a z-gradient TXI cryoprobe or on a Bruker DRX-500 with a xyz-gradient TXI probe. The 1D and 2D spectra were recorded at 273 K. The 2D NOESY spectra of the imino region were recorded with 50 or 300 ms mixing times. A combination of Watergate and Jump and Return sequences was used for water suppression [9]. 15N HSQC spectra were acquired on the 15N labeled samples to confirm the base-type assignments [10]. Proton and nitrogen chemical shifts were calibrated against TSP (Trimethylsilylpropionic acid).
The NMR protein samples typically contained 0.5–0.8 mM protein (except for RRM1,2 that contains about 0.1 mM protein) in 50 mM sodium phosphate (pH = 4), 50 mM sodium chloride, 50–100 mM imidazole, 1 mM EDTA, and an anti protease cocktail. 15N HSQC spectra were acquired on the 15N labeled protein samples at 293 K on a Bruker DRX-600 with a z-gradient TXI cryoprobe.
All the 2D spectra were collected in a phase-sensitive mode via the STATES-TPPI method, and processed accordingly using XwinNMR software.
3 Results and discussion
3.1 Selection of minimal RNA sequences and protein constructs as models for the NMR analysis
Several RNA molecules, containing the pESS2 sequence (shaded in Fig. 1), have been synthesized. The goal was to shorten the long irregular SLS2 stem-loop, whose secondary structure may influence its activity, to a size suitable for a NMR study. Four RNA molecules were designed and synthesized as shown in Fig. 1 in comparison with the original HIV Bru sequence. Two stem-loops (A3_1 and A3_2) were fist engineered, which are essentially similar to the original sequence, except for additional GC base-pairs added for transcription purposes and additional stabilization of the stem-loop. In addition and for resonance assignment purposes, two shorter sequences corresponding to the apical loop with a short stem (STEP1) and with a stem, where the bulged residues were omitted (STEP2) were also synthesized.
The heterogeneous ribonucleoprotein H (hnRNP H) is a multidomain protein that contains three RRMs and low complexity glycine rich domains (Fig. 2). This 50-kD protein is at the upper size limit for extensive NMR studies, but it has been shown that the first two RRM domains are responsible for the binding to ESS2 and the glycine rich domains are probably involved in protein–protein recognition and could help the recruitment of additional trans-factors. We first decided to work on a construct encompassing the RNA binding domains 1 and 2, leading to a 25-kD protein. To help in the assignment of this still large protein, we also produced the isolated domains 1 and 2. One of the problems of this type of strategy is the proper delineation of the domains and the transferability of the assignments to the parent protein. The first problem was addressed by a bioinformatics strategy using a combination of secondary structure prediction algorithms (HCA and the package of softwares available through the npsa server [6]), alignment strategies (SMART, Pfam) or database searches (PSI-BLAST). A large consensus of the results obtained with the various methods and programs allows to limit the first RRM between residues 10 and 92 and the second between residue 110 and 190, both with a βαββαβ fold prediction.

Designed hnRNP H constructs and their 600 MHz HSQC spectra. (A) Pfam [11] schematic representation of hnRNP H (SwissProt entry P31943) and of the various constructs (RRM1,2 corresponds to the first two RRM domains, RRM1 and RRM2 to the first and second RRM domain, respectively). The sequence numbering corresponds to the hnRNP H sequence. (B) Superposition of the 600-MHz HSQC spectra at of RRM1 (green), RRM2 (red) and RRM1,2 (blue) at 298 K. (C) Zoom of the glycine region of the superimposed HSQC spectra. (D) Zoom of the asparagine/glutamine sidechain region of the superimposed HSQC spectra. Masquer
Designed hnRNP H constructs and their 600 MHz HSQC spectra. (A) Pfam [11] schematic representation of hnRNP H (SwissProt entry P31943) and of the various constructs (RRM1,2 corresponds to the first two RRM domains, RRM1 and RRM2 to the first ... Lire la suite
3.2 RNA secondary structure determination
Standard methods were used to deduce the secondary structure and global fold from the assignment of the exchangeable protons [10].
Initial assignments of A3_1 and A3_2 were based on standard NOESY walk analysis (Fig. 3). It was straightforward to assign base-paired NH resonances from the predicted Watson–Crick pairs of the stem. The GC and AU pairs were identified from the imino–amino and the imino-H2 cross-peaks, respectively. Three GU base-pairs were identified from the typical imino–imino cross-peaks within the pair observed at short mixing times and were identified to base-pairs 4, 5 and 7, based on the sequential connectivities. The folding as a stem-loop with a two base-pair bulge was clearly demonstrated from the sequential connectivities as shown for A3_1 in Fig. 3. Six additional resonances corresponding to protected residues, three of them being base-paired and three not, were also present and would correspond to protected residues within the apical loop, in agreement with the enzymatic and chemical foot-print experiments (data not shown). In addition, a minor set of resonances was also identified in the spectrum. To help in the assignment of these resonances, two shorter sequences were also engineered, that correspond to the upper stem (STEP1) and a sequence that does not present the bulge of the original sequence (STEP2). NMR experiments unambiguously demonstrated that both sequences fold as double stranded dimers (data not shown). As such, these shorter sequences proved of no use in assigning the additional resonances of the major form and to give information on the extensive base-pairing of the loop. But several resonances matched the low field resonances of the minor set of resonances, insuring that the initial sequence folds mainly as a stem loop in our conditions and that the minor form is the corresponding double stranded dimer. These preliminary NMR experiments demonstrate that the engineered A3_1 RNA fragment adopts the expected bulged hairpin structure, probably with additional extensive folding within the loop, as predicted for the native sequence in SLS2 and represents a proper model for the interaction studies by NMR. The NMR characteristics of A3_2 are essentially similar. The imino protons assignment paves the way for preliminary interaction studies.

Imino protons attribution of RNA A3_1. (A) 500-MHz NOESY spectrum at 273 K. (a) Three different regions of a NOESY spectrum with 300 ms mixing time. (b) Imino–imino region of NOESY spectrum at 50 ms mixing time. (b) 500 MHz 1D spectrum at 273 K. Arrows indicate unidentified imino protons. Asterisks label imino protons that belong to the duplex form. (c) Resonance assignment. Sequential connectivities are shown with different colors.
3.3 Protein production and preliminary NMR studies
Various strategies were tested in order to achieve an efficient production of the three recombinant proteins. The crucial factor in our case proved to be a proper control of the transcriptional regulation. For large-scale gene expression, we performed cell growth to high density (estimated from A600 nm) and minimal promoter activity, followed by induction of the promoter. But in the present case, special care has to be taken that cell growth density never becomes higher than A600 nm = 0.8–1.0 before induction. With greater values, incompletely repressed expression occurs and usually causes plasmid instability, and a severe drop in recombinant protein production (data not shown). This point proved crucial for all our constructs. Under the above conditions, a high level of production of unlabelled and labeled samples was repeatedly achieved (up to 10 mg/l).
The binding specificity of the RRM1,2 construct on pESS2 was tested at MAEM using gel shift methods (data not shown).
Further interaction studies using NMR spectroscopy need protein attribution. Resonance assignment of proteins above 20 kD remains a tedious task. To facilitate RRM1,2 resonance assignment, we decided to study each RRM domain separately using our RRM1 and RRM2 constructs. This is only relevant if each domain folds separately, keeps the same fold in the RRM1,2 bimodule and if the two modules do not extensively interact in the latter. To ensure this hypothesis, we superimposed the three HSQC spectra (Fig. 2). The HSQC spectrum of RRM1,2 is almost identical to the sum of the individual HSQC spectra of RRM1 and RRM2. This is illustrated in the two zooms of the figure. The first zoom corresponds to part of the glycine region. Glycine residues are homogeneously distributed along the primary sequence and we can see that correlation peaks match very well. Also convincing is zoom 2, focused on several arginine and glutamine side-chains departing from the standard chemical shifts of free side-chains. Here again, peaks are superimposable, which means that not only the main chain but also the side-chains seem to adopt the same conformation in the isolated modules and in the RRM1,2 bimodule. In conclusion, it is now clear that the RRM1,2 construct behaves as two independent domains in the absence of the target RNA and that the resonance assignment of the RRM1 and RRM2 domains will ease the necessary resonance assignment for the RRM1,2, opening avenues for the interaction studies with the engineered RNA targets.
Acknowledgements
We thank Houda Hallay (MAEM, Nancy) for providing the plasmid for the RRM1,2 construct. S.C. was supported by a grant from the ‘Agence nationale de la recherche sur le sida’ (ANRS).