

1 Introduction
Lanthanide complexes have been shown to function as UV light conversion devices [1], resulting in a strong red luminescence. Their cryptates also present interesting perspectives as luminescent labels [2]. New developments involve the design of novel ligands in order to further improve the photophysical properties. Pure organic species like organic light-emitting diodes (OLEDs) have also attracted considerable scientific and industrial attention [3] due to their potential application in large area flat-panel displays [4]. For full-color applications, it is necessary to have a set of red, green, and blue emitters with sufficiently high luminous efficiency and proper chromaticity. Actually, organic materials for green and blue OLEDs with high luminescence have largely been developed [5]. Polyfluorenes are leading candidates for blue OLEDs [6] and are expected to be part of the full-color polymer displays. The corresponding organic materials for red electroluminescence (EL) are still rare [7]. Red light emission is usually achieved by doping red dyes into host materials with a large gap [8]. Red dyes, such as porphyrins, are organic molecules with reasonable fluorescence efficiency and good thermal stability. Thus, a conjugated polymer, poly(9,9-dioctylfluorene), doped with a tetraphenylporphyrin molecule [9] dispersed into host matrix, has been successfully used as a light-emitting diode. A series of star-shaped porphyrins bearing pendant oligofluorene arms, recently reported by Bo et al. [10,11], offer a second possibility. In this example, the porphyrin and the polyfluorene groups are conjugated in order to achieve through-bond energy transfer between the fluorene and the porphyrin core. The rate of through-bond energy transfer is normally a hundred times faster than that of through-space energy transfer (Förster) [12,13]. We have herein considered the design of new building blocks based on fluorenylporphyrins with the idea to obtain red emitting organic materials after polymerization.
We have previously reported the synthesis of a free macrocycle possessing four pendant fluorene arms at the meso-positions and studied the catalytical properties of the ruthenium (II) complex, either as a homogenous catalyst or as a polymer heterogeneous material [14,15]. Very efficient catalysts for 2,3 sigmatropic reactions or classic cyclopropanation were thus developed which could be easily recovered and re-used [14,16,17]. We now want to focus on the photophysical properties of such porphyrins, including the fluorenone analogue. The tetraphenylporphyrin macrocycle (H2TPP) will presently be used as a reference. These building blocks, exhibit very efficient energy transfer through bonds. Since metallation often makes the macrocycle more rigid and symmetrical, the zinc complexes have been prepared [18] and their photophysical properties investigated.
2 Results and discussion
2.1 Synthesis of porphyrins and metalloporphyrins
Meso-tetrafluorenylporphyrin 1 was prepared by condensation of pyrrole and 2-fluorenecarboxaldehyde as previously reported (Fig. 1a) [14]. Compound 2 was synthesized by adding macrocycle 1 to a mixture of heptane under very basic conditions (NaOH in water) in presence of a phase-transfer reagent (Aliquat 336). Oxidation gave the tetrafluorenone macrocycle 2 with a good yield (68%) (Fig. 1a).
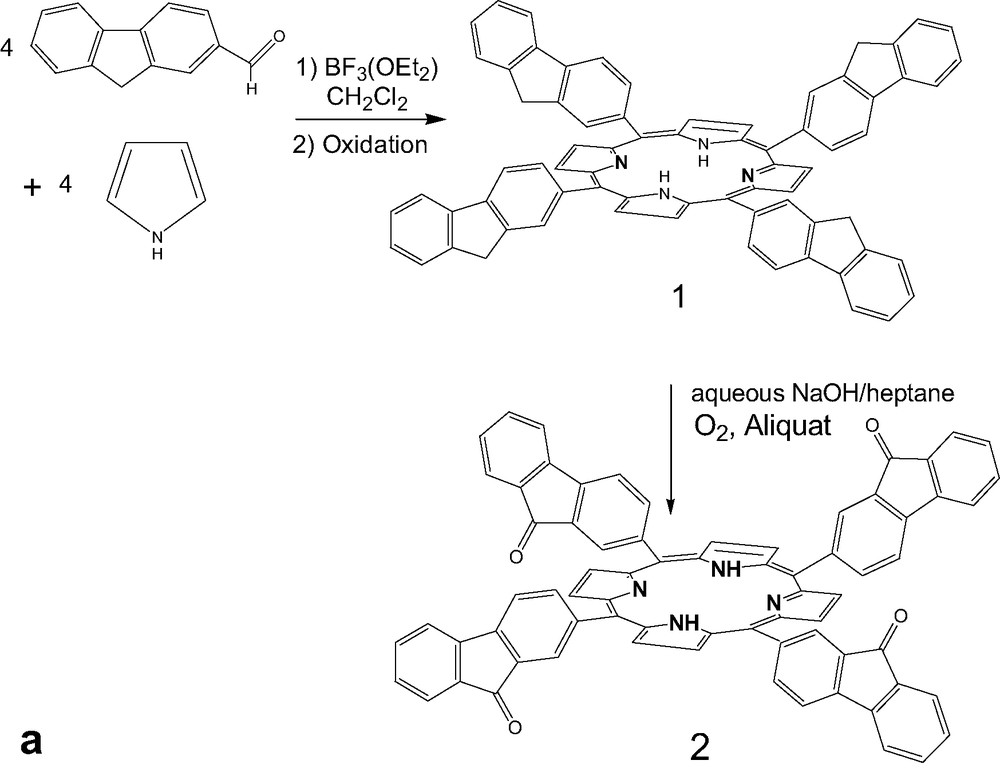
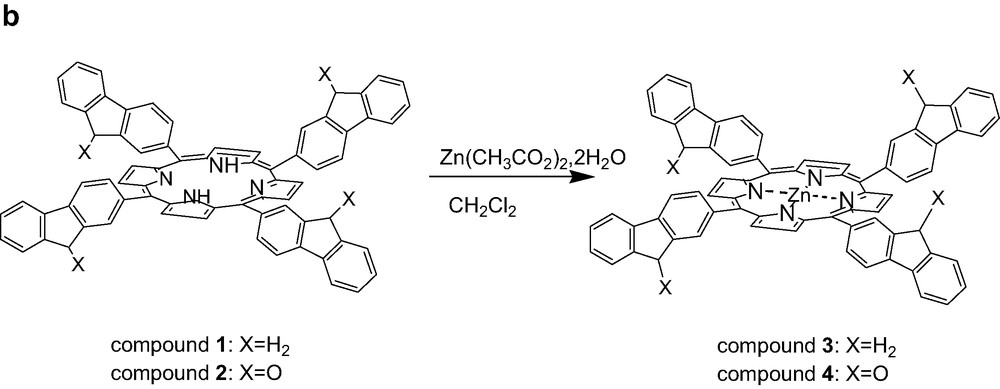
(a) Synthesis of tetrafluorenoneporphyrins 2, (b) Preparation of zinc complexes of ligand 1 and 2.
The zinc porphyrin monomers, 3 and 4 (Fig. 1b) were prepared by treatment of free ligands 1 and 2 with (Zn(CH3CO2)2·2H2O) in dichloromethane at room temperature [19]. Whereas, the reaction time is only 12 h for complete metallation in case of the phenyl substituent (5), 48 h are necessary for 1. This could be attributed to the steric effect of the bulky meso-fluorene groups. In contrast, for the metallation of free-base tetrafluorenoneporphyrin (2), the reaction time is only 3 h, as revealed by UV-monitoring. Probably, the strong withdrawing electron fluorenone arm is the major effect in this case.
Surprisingly, the proton NMR signals obtained in deuterated chloroform for zinc complex 4, were very broad (Fig. 2b). This was not observed for the zinc complex 3. Aggregation is often observed with porphyrins and metalloporphyrins bearing strongly electron withdrawing groups at either the meso or β position [20]. Presently, the [ZnTFOP] complex 4 has two potentially interacting functional sites: C=O and Zn(II) [21]. We could hinder π-staking in the case of these zinc complexes, by making use of the supplementary coordination sites at the apical positions, with ligand like pyridine. Accordingly, addition of a slight quantity of pyridine renders the 1H NMR spectrum immediately well resolved (Fig. 2c). Tamiaki et al. [22] have indeed shown earlier that oligomeric forms can disappear with addition of a coordinating ligand like THF, giving monomer species. Ligation of pyridine ring to zinc porphyrin is intrinsically weak (association constant 10−3 M−1) and, in solution, coordination oligomers are generally in equilibrium with monomers [23]. A large amount of monomers is therefore expected at the low concentrations used for photophysical measurements (ca. 10−6 M), even in the absence of pyridine.

1H NMR spectra of:
(a) H2TFOP (2), (b) [ZnTFOP] (4), (c) [ZnTFOP + pyridine].
2.2 Photophysical properties of free macrocyclic ligand
Energy transfer from fluorene groups to porphyrin core was anticipated to favor red light emission. Therefore, the photophysical properties of the free ligands, H2TFP and H2TFOP, in dichloromethane solution were studied, using H2TPP as a reference.
2.2.1 Electronic spectra
The porphyrin skeleton has an extended π-conjugation system with 24-π electrons leading to a wide range of wavelengths for light absorption. The bands in different spectral regions are denoted as Q, B, N, L and M. The most representative of these, are the Q bands, the weak absorptions in the visible region, and the Soret band B, more intense absorption occurring in the near UV region. The other bands labeled N, L and M appear as weak absorption in the UV region [24]. Variations of substituents on the porphyrin ring often cause minor change to the intensities and wavelengths of these absorptions [25].
Actually, it is known that the meso-aryl groups are not orthogonal to the porphyrin plane, but slightly tilted [26], allowing the meso group to develop some conjugative interaction with the main porphyrin framework. Thus, in spite of the large dihedral angle between the plane of the phenyl ring and the porphyrin π system in the H2TPP series, conjugation exists to some extent and is affected by the substituents on the phenyl ring. This conjugative interaction of the aromatic meso-substituents with HOMO level a2u can explain the slight red shift observed for the fluorene derivative 1 which exhibits a Soret band at 425 nm (Fig. 3), in comparison to the absorption of 5 (417 nm).
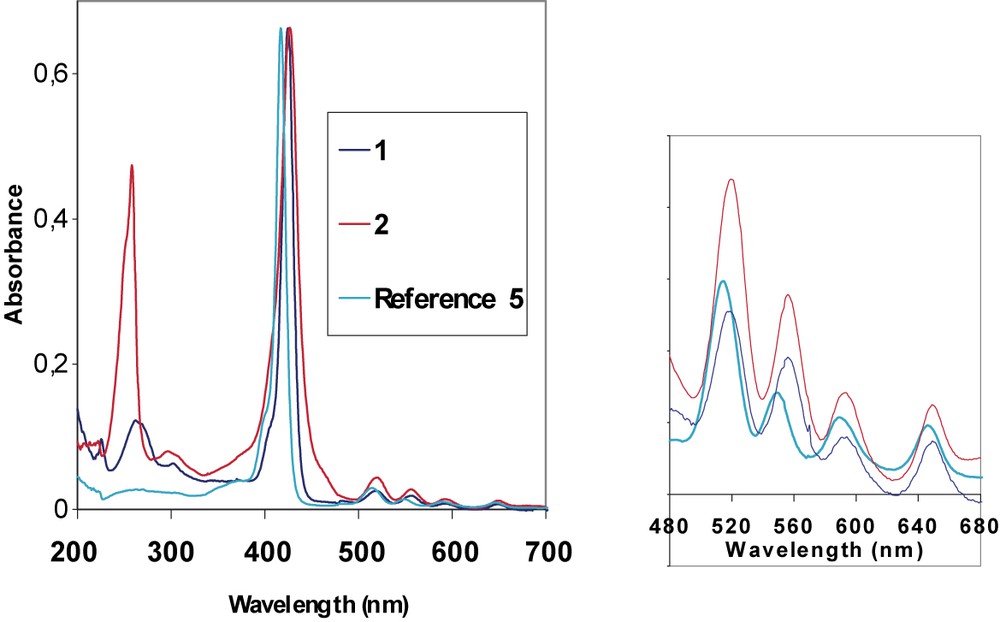
UV–visible absorption spectra for compounds 1 and 2 compared to reference 5.
In the visible spectrum, the macrocycle 1 exhibits an intense B(0,0) band, together with four weak Q-bands between 500 and 700 nm. An additional broad band corresponding to the fluorene absorption is also observed in the UV range (268 nm), blue shifted from ca. 40 nm in comparison to fluorene (301 nm).
The UV–visible spectrum of 2 exhibits an intense Soret band with a maximum absorption around 427 nm (Fig. 3). This band is also red shifted (10 nm) compared to 5, as well as the Q bands. The fluorenone arms absorb in the UV range with a maximum absorption peak at 258 nm, this peak is remarkably sharper and stronger than the corresponding fluorene absorption peak (Fig. 3).
2.2.2 Luminescence studies
The fluorescence of the free macrocycles 1 and 2 was studied in dichloromethane (using dilute solution in order to avoid self-quenching), while the measurements for the quantum yield were done in toluene to allow comparison with reported references.
The emission spectrum of compound 1, after excitation in the fluorene absorption (262 nm), in the Soret band (425 nm) or in the highest Qy(0,1) transition (519 nm) reveals a strong red fluorescence with peak maximum at 662 nm and a weak shoulder at 726 nm (Fig. 4a). Intensity of luminescence increases following λ262 < λ519 < λ425.
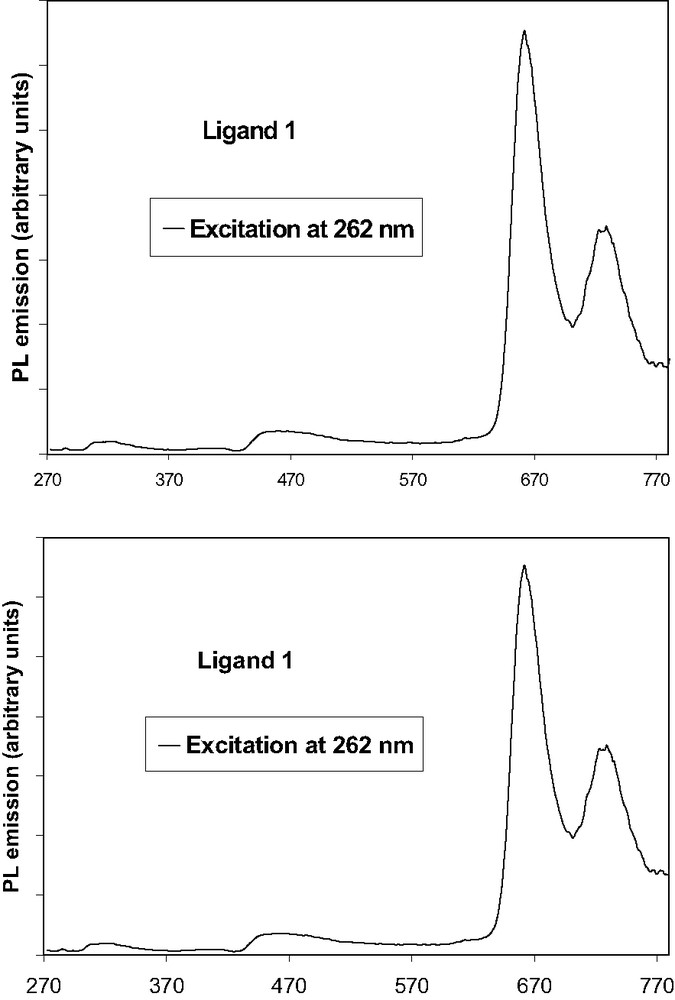
(a) Photoluminescence spectrum of free ligand 1, (b) Photoluminescence spectrum of free ligand 2.
The excitation spectrum obtained after exciting in the strongest emission band at 661 nm, reveals the Soret band, the three first Q bands (Qy(0,1), Qy(0,0) and Qx(0,1)), and the fluorene. This indicates that excitation over all the 200–650-nm region leads to the population of the fluorescent excited state of the porphyrin, except for the fluorene chromophore involved in the UV absorption process.
There is a good energy transfer between fluorene and porphyrin, since almost no residual emission of the fluorene is observed in comparison to the porphyrin emission (Fig. 4a). Thus, almost all the absorbed energy by the antennae is reemitted by the porphyrin rings. The remaining emission from fluorenes in compound 1 (~300 nm) accounts for the competition between the direct emission from fluorenes and energy transfer to the porphyrin macrocycle.
The low Stokes shift observed between the lowest absorption at 651 nm and the fluorescence band 661 nm, indicates that the distortion occurring in going from the ground state to the fluorescent excited state is not important.
The emission spectrum of compound 2 after excitation> in the fluorenone arms (258 nm), in the Soret band (427 nm) or in the highest energy Qy(0,1) transition (519 nm) also reveals strong red light emission (Fig. 4b). Intensity of luminescence increases following λ260 ≈ λ519 < λ427. So there is also an efficient energy transfer between antennae and porphyrin for compound 2, as illustrated by the spectrum between 260 and 800 nm (Fig. 4b). Again, there is evidence of almost no residual emission of the fluorenone units (~300 nm) in comparison to the porphyrin emission, so quite all the absorbed energy by the antennae is reemitted by the porphyrin. Finally, the Stokes-shifts increase only slightly when the fluorene (10 nm) is replaced by fluorenone (12 nm) (compound 2).
2.3 Photophysical properties of the zinc complexes
The distortion due to general flexible character of the free macrocycles 1 and 2, causes a significant break up in conjugation. Accordingly, there is an alteration of π-energy levels, including a destabilization of the HOMO, and some stabilization of the LUMO. The net result is to reduce further the HOMO–LUMO gap. Previous theoretical calculations (INDO/s calculations) are in agreement with these observations [27]. By introducing a metal in the central cavity, the optimal conjugation should therefore be restored. The porphyrin macrocycles 1 and 2 were metallated with zinc to afford more rigid luminophores 3 and 4. We studied the photophysical properties of these new Zn2+ complexes 3 and 4 in dichloromethane solutions. The Zn(II) porphyrin complexes exhibit characteristic change in the electronic spectra compared to the free-base porphyrins.
2.3.1 Electronic spectra
The absorption spectra of Zn(II) porphyrins display a single Q band absorption which is a combination of the QX and QY bands due to the D4h symmetry of metal porphyrins instead of the D2h symmetry of free-base porphyrins. Thus, the zinc(II) complexes have two discernible Q bands (Q(0,1) and Q(0,0)) [24,28,29]. In the visible spectra, 3 and 4 exhibit an intense B band together with two weak Q-bands. An additional broad band corresponding to π–π* absorption of fluorene (3) or fluorenone (4) is still observed in UV range. Notably, the UV absorption is particularly strong for the fluorenone derivative 4 (Fig. 5). The π–π* absorption in the UV range at 260 nm, for fluorenone is almost as big as the Soret band at 431 nm, for this compound, the absorption is notably exalted.
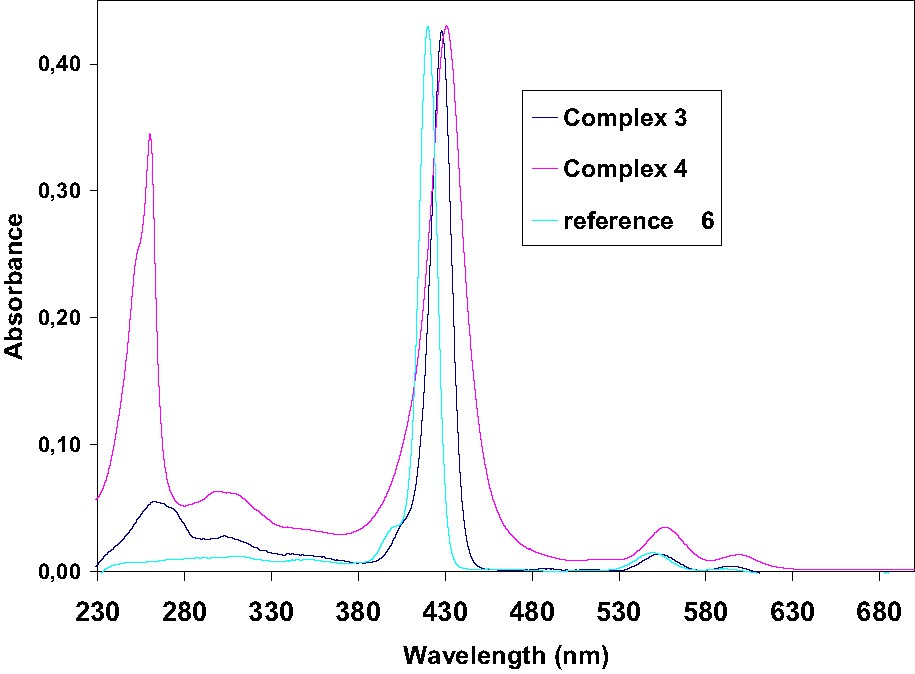
UV–visible spectra of zinc complexes 3 and 4 compared to reference 6.
The absorption spectrum of complex 3 in CH2Cl2 displays a Soret band at 428 nm and the two Q bands: (Q(0,1) and Q(0,0)) at 551 and 594 nm, respectively. On the other hand, the spectrum of complex 4 exhibited an intense Soret band with a maximum absorption around 431 nm and the two Q bands at 556 and 599 nm. In both case, these bands are red shifted compared to the reference [ZnTPP] compound 6 (421, 550 and 585 nm, respectively). Reduction of the distortion of the free macrocycle by metallation increases only slightly the red-shift of the Soret band in comparison to the non metallated analogues 1 and 2 (ca. 3 nm).
2.3.2 Luminescence studies
Room temperature fluorescence spectrum of zinc(II) complex 4 is shown in Fig. 6. The emission spectrum consists of three bands. The weak band near 720 nm is assigned as Q(2,0) and actually appears as an extended tail on the Q(1,0) band. The major difference between the zinc complexes [ZnTPP], [ZnTFP] and [ZnTFOP], compared to their corresponding free-base H2TPP, H2TFP and H2TFOP, is the remarkable hypsochromic shift of ~50 nm for the strongest emission band Q(1,0) and 70 nm for the second Q(0,0), due to the metal coordination.
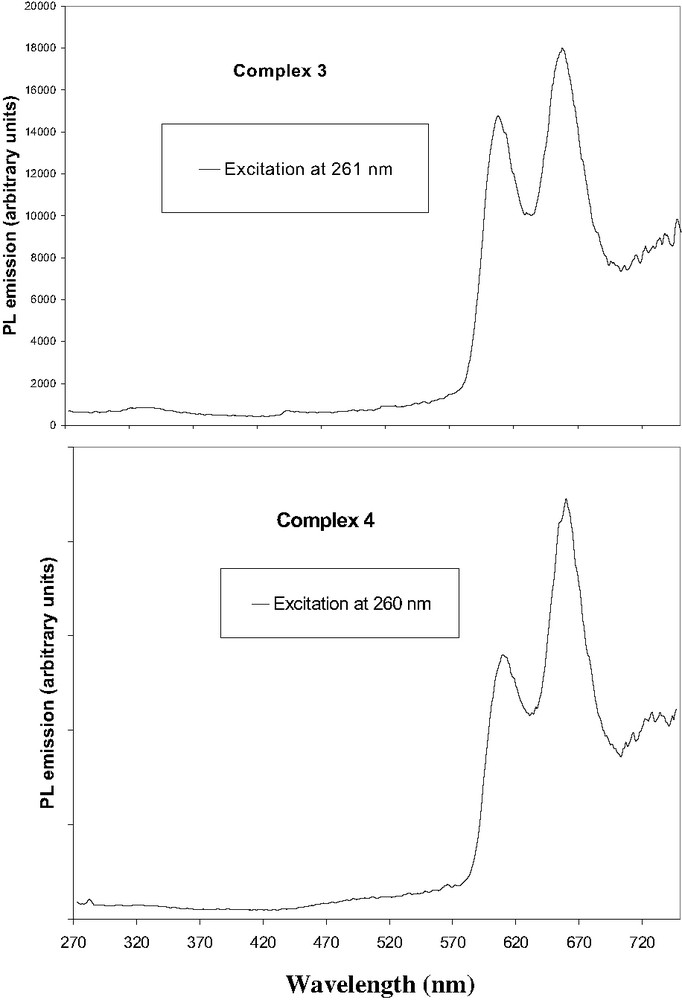
(a) Photoluminescence spectrum of zinc complex 3, (b) Photoluminescence spectrum of zinc complex 4.
Excitation of compound 3, at the π–π* absorption of the fluorene, the Soret band and the first Q band gave a fluorescence with peak maxima at 609, 659 nm and a very weak at 725 nm. Thus, the difference in wavelength compared to the corresponding free ligand 1 is 53 nm for Q(1,0) and 68 nm for the Q(0,0). For complex 6, a narrow peak at 610 nm and weak band at 660 nm are attributed to the excited triplet states of the Q-bands.
As observed on the spectrum between 270 and 720 nm, there is evidence of almost no residual emission of the fluorenone units. This contrasts slightly with the emission of the free ligand.
We next studied the complex 4 in dichloromethane, by excitation at the same wavelengths, i.e. in the π–π* of the fluorenone (260 nm), the Soret band (431 nm) and first Q band (556 nm). Fluorescence peaks with maximum at 614 nm, 660 and 725 nm were observed (Fig. 6). The emission profiles of [ZnTFP] and of [ZnTFOP] are nearly identical, except the weak band near 725 nm assigned as Q(2,0) which actually appears stronger for complex [ZnTFP].
For ZnTFP, when monitoring the emission wavelength at 610 and 660 nm, the excitation spectrum, exhibits an intense Soret band at 428 nm and the Q(1,0) and Q(0,0) bands at 551 and 594 nm, respectively, which roughly correspond to the ground state absorption spectrum. This indicates that excitation over all the 240–700 nm region leads efficiently to the population of the fluorescent excited state of the porphyrin except for the fluorene chromophore involved in the UV absorption process.
A self-quenching experiment on the emission of [ZnTFP] and [ZnTFOP] was performed in the concentration range 1 × 10−6 to 1 × 10−5 M in CH2Cl2 at 298 K, and a detectable quenching effect was found at ~10−5 M.
Emission spectra of [ZnTPP], [ZnTFP] and [ZnTFOP] by excitation at antennae and Q band were then studied. For complex 3 and reference 6, the emission after exciting at 260 nm, is very weak in comparison to the first Q band (ratio is 6.5 and 4.2). This was also observed for the corresponding free ligands. In contrast, for 4, the luminescence is similar when exciting the antennae or to the first Q band (ratio is 1.3) confirming an efficient energy transfer between fluorenone and porphyrin. There is no evidence of residual emission of the fluorenone units (Fig. 6).
Again, slight Stokes-shifts are observed for complexes 3 and 4. Actually, these shifts are similar for both compounds (15 nm).
2.4 Fluorescence quantum yields
The fluorescence quantum yields of free ligands 1 and 2 were determined by comparing with a calibration standard of 5 in degassed toluene solution with a fluorescence quantum yield of 0.12 [30], preferentially to a benzene solution with a fluorescence quantum yield of 0.13 [7] (in this case different refractive indices of the solvents used in the standard and sample must be corrected) [31]. All measurements were made under argon atmosphere to limit photo-oxidative degradation.
The quantum yield was calculated from the following equation:
In the above expression, ΦS is the fluorescent quantum yield of the new compound, F is the integration of the emission intensities, n is the refraction index of the solution, and A is the absorbance of the solution at the exciting wavelength. The subscripts I and S denote the reference and unknown samples, respectively [32].
Values of quantum yield of free ligands 1 and 2 are reported in Table 1. The corresponding zinc complexes 3 and 4, showing less intense fluorescence in solution at room temperature, are also reported.
Quantum yield (%) of free macrocycles and zinc complexes
| Quantum yield (%) of free macrocycles | |||
| Compounds | H2TPP(5) | H2TFP(1) | H2TFOP(2) |
| λex (nm) | 420a | 427 | 431 |
| λem (nm) | 653 and 721 | 661 and 726 | 661 and 727 |
| Quantum yield (%) | 12a | 22 | 25 |
| Quantum yield (%) of zinc complexes | |||
| Compounds | ZnTPP(6) | ZnTFP(3) | ZnTFOP(4) |
| λex (nm) | 423b | 430 | 434 |
| λem (nm) | 597 and 646 | 609 and 660 | 614 and 661 |
| Quantum yield (%) | 3.3b | 5.0 | 4.9 |
Compound 1 presents an interesting luminescence quantum yield (22%), which is higher than that of the reference compound 5 (12%). Compound 2 possesses also a high luminescence quantum yield (25%), slightly higher than the parent compound 1.
Complexes 3 and 4 have low luminescence quantum yield (around 5%), which is higher than this of the reference compound 6 (3.3%).
3 Conclusion
In summary, we have synthesized and characterized a new porphyrin 2, bearing fluorenone pendant arms at the meso-positions starting from free macrocycle tetrafluorenylporphyrin 1, as well as the corresponding zinc complexes 3 and 4.
We have shown that free porphyrins 1 and 2 emit red light after UV–visible irradiation, as well as the corresponding zinc complexes 3 and 4. As expected, the compounds deliver good red chromaticity. The conjugation system of these porphyrins with pendant arms is enhanced compared to 5, resulting in a longer wavelength of the absorption and emission peaks. The energy transfer process from the fluorene or fluorenone arms to the porphyrin cycle is apparently very efficient. In comparison to the reference macrocyclic 5, the luminescence properties are doubled, as revealed by the measurement of the fluorescence quantum yields that range from 0.22 (for 1) to 0.25 (for 2), values much higher than for many other porphyrins [33]. The fluorescence of the Zn(II) complexes 3 and 4 is however less intense than 1 and 2, but higher than the corresponding zinc complex 6.
These molecules might therefore be of high interest to make luminescent materials. These promising results prompt us to design now ligands incorporating more than four antennae in order to achieve higher luminescence, and to explore oxidative electropolymerization of these new compounds.
4 Experimental section
4.1 General procedures
All reactions were performed under argon and were magnetically stirred. Solvents were distilled from appropriate drying agent prior to use, CH2Cl2 from CaH2, CHCl3 from P2O5 and all other solvents were HPLC grade. Commercially available reagents were used without further purification unless otherwise stated.
All reactions were monitored by TLC with Merck pre-coated aluminum foil sheets (silica gel 60 with fluorescent indicator UV254). Compounds were visualized with UV light at 254 and 365 nm. Column chromatography was carried out using silica gel from Merck (0.063–0.200 mm). 1H NMR and 13C NMR in CDCl3 were recorded using Bruker 200 DPX, 300 DPX and 500 DPX spectrometers. The chemical shifts are referenced to internal TMS. The assignments were performed by 2D NMR experiments: Correlation Spectroscopy (COSY), Heteronuclear Multiple Bond Correlation (HMBC) and Heteronuclear Multiple Quantum Coherence (HMQC). High-resolution mass spectra were recorded on a ZabSpec TOF Micromass spectrometer in FAB mode or ESI positive mode at the CRMPO. IR spectra were recorded on Bruker IFS 28, using KBr pellets or in dichloromethane solution. UV spectra were recorded on UVIKON XL from Biotek instruments. PL emission was recorded on a Photon Technology International (PTI) apparatus coupled on an 814 Photomultiplier Detection System, Lamp Power Supply 220B and MD-5020. Aliquat 336, pyrrole and 2-fluorenecarboxaldehyde were purchased from Aldrich and were used as received. TFP = tetrafluorenylporphyrin dianion, TPP tetraphenylporphyrin dianion, TFOP = tetrafluorenoneporphyrin dianion.
4.2 Porphyrin synthesis
4.2.1 Meso-tetrakis-5,10,15,20-(fluoren-2-yl)-porphyrin 1
It was synthesized as previously described in [14].
4.2.2 Meso-tetrakis-5,10,15,20-(fluorenone)-porphyrin 2
It was synthesized by oxidation of precursor 1, as described below. One equivalent (0.1 mmol) of compound 1, was dissolved in 40 ml of heptane. Aqueous NaOH (30%, 20 ml) is carefully added, followed by addition of 20 drops of Aliquat 336 (tricaprylmethylammonium chloride). The mixture is stirred vigorously for 4 days. Reaction progress is monitored by TLC, spotting directly from the organic layer. Then, the dark brown heptane solution was separated and concentrated. The dark brown crude is chromatographed on silica gel column with a dichloromethane/heptane (80:20) mixture to dichloromethane. The red–brown crystalline compound 2, meso-tetrakis-5,10,15,20-fluorenoneporphyrin was obtained with a yield of 68%.
This new compound 2, was characterized by UV–visible, IR, NMR, microanalysis and mass spectrometry. The hydrogen and carbon atom-labeling scheme for the porphyrin ligands is shown below (Fig. 7).
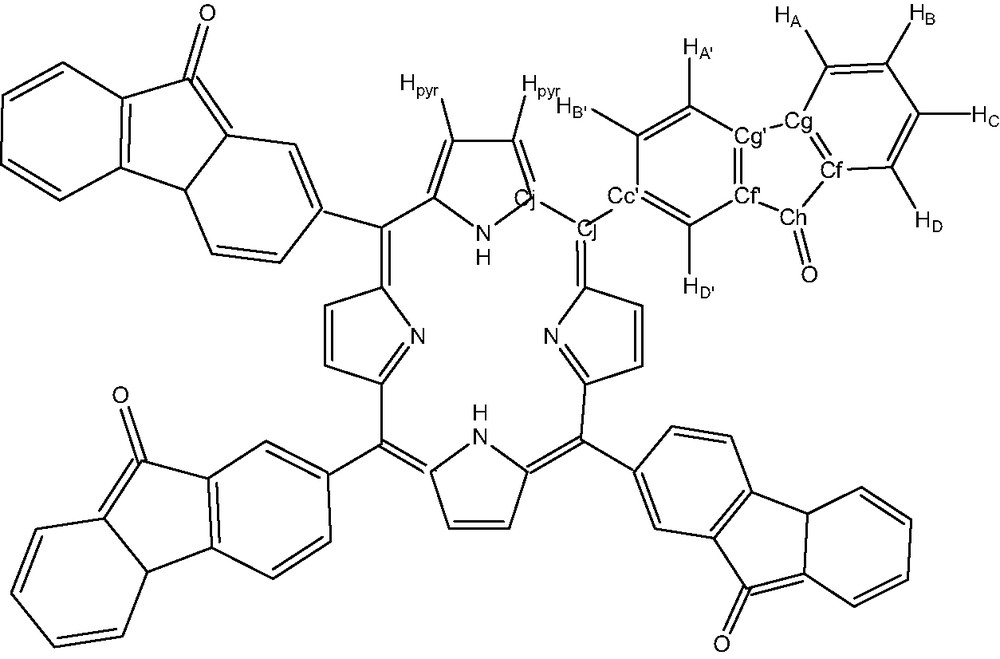
Free ligand tetrafluorenoneporphyrin: compound 2.
Characterization by NMR (DEPT 135, proton decoupling, NOE difference, HMQC and HMBC, high and low temperature) of free ligand H2TFOP (2):
- ● 1H NMR (CDCl3): 8.98 (s, 8H, pyrrole), 8.56 (s, 4H, HD′), 8.36 (d, 3JHH = 7.3 Hz, 4H, HB′), 7.98 (d, 3JHH = 7.2 Hz, 4H, HA′), 7.86 (d, 3JHH = 6.2 Hz, 4H, HD), 7.83 (d, 3JHH = 6.8 Hz, 4H, HA), 7.69 (t, 3JHH = 7.7 Hz, 4H, HB), 7.48 (t, 3JHH = 7.3 Hz, 4H, HC), –2.72 (s, 2H, NH).
- ● 13C NMR (CDCl3): 194.23 (CE), 146.69 (CJ), 144.42 (CG), 144.05 (CG′), 143.05 (CC′), 140.28 (CB′), 135.01 (CF), 134.99 (CB), 132.98 (CF′), 131.20 (Cpyrrole), 129.55 (CD′, CC), 124.58 (CD), 120.85 (CA), 119.38 (CI), 118.88 (CA′).
- ● UV–visible (in CH2Cl2): 258 nm (fluorenone, ε258 = 335 128), 427 nm (Soret band, ε427 = 506 282), 519 nm (Q band, ε519 = 23 974), 556 nm (Q band, ε556 = 18 589), 593 nm (Q band, ε593 = 9448), 649 nm (Q band, ε649 = 8333). IR: 1715 cm−1 (CO band), 1613 cm−1 (fluorenone), 1601 cm−1, (fluorenone), 1455 cm−1 (fluorenone), 1107, and 736 cm−1. Analysis: calcd for C72H38N4O4·2 CH2Cl2 C: 74.50, H: 3.55, N: 4.70, found C: 74.24, H: 3.84, N: 4.58. MS (ESI): calcd for C72H38N4O4: 1023.2971 [MH]+, found: 1023.2978 [MH]+.
4.3 Porphyrin zinc complexes synthesis
4.3.1 Meso-tetrakis-5,10,15,20-(fluoren-2-yl)porphyrinato zinc(II) 3
Free-base porphyrin 1 (100 mg, 0.10 mmol) was dissolved in 20 ml of distilled dichloromethane. Zinc acetate dihydrate (108 mg, 0.49 mmol) is added in one portion to the stirred solution. The mixture was stirred vigorously for 48 h at room temperature. Reaction progress is monitored by TLC, spotting directly from the organic layer. Then, the solvent was removed, and the black red residue was chromatographed on silica gel column with a MeOH/CH2Cl2 mixture (2–10% MeOH). Compound 3, was obtained as a dark-red color powder in a yield of 87%. This new porphyrin complex (3) was characterized by NMR, UV–visible, IR, microanalysis and mass spectrometry.
1H NMR (CDCl3): 8.97 (s, 8H, pyrrole), 8.42 (s, 4H, HD′), 8.28 (d, 3JHH = 8.2 Hz, 4H, HB′), 8.20 (d, 3JHH = 7.6 Hz, 4H, HA′), 8.10 (d, 3JHH = 6.6 Hz, 4H, HA), 7.72 (d, 3JHH = 6.8 Hz, 4H, HD), 7.56 (t, 3JHH = 5.7 Hz, 4H, HB), 7.46 (t, 3JHH = 6.8 Hz, 4H, HC), 4.23 (s, 8H, HE).UV–visible (CH2Cl2): 263 nm (fluorene, ε263 = 81 323), 428 nm (Soret band, ε428 = 627 058), 552 nm (Q band, ε552 = 20 735), 594 nm (Q band, ε594 = 6618). IR: 2961, 2924, 2853, 1464, 1452, 1415, 1396, 1346, 1261, 1097, 1022, 800 cm−1. Analysis: calcd for C72H44N4Zn.2CH2Cl2·3CH3OH C: 71.33, H: 4.66, N: 4.32, found C: 71.17, H: 5.08, N: 3.90 MS (ESI): calcd for C72H44N4Zn: 1030.4703 [MH]+, found: 1030.470 [MH]+.
4.3.2 Meso-tetrakis-5,10,15,20-(fluorenone)porphyrinato zinc(II) 4
Free-base porphyrin 2 (100 mg, 0.10 mmol) was dissolved in 20 ml of distilled dichloromethane. Zinc acetate (108 mg, 0.49 mmol) was added in one portion to the stirred solution. The mixture was stirred for 3 hours at room temperature. Then, the solvent was removed, and the black green residue was chromatographed on silica gel column with a MeOH/CH2Cl2 mixture (2–10% MeOH). Compound 4, was obtained as a dark green color powder in a yield of 90%. Surprisingly, the proton NMR spectrum is very large and broad in deuterated chloroform. By adding progressively pyridine, the spectrum becomes less complex and gives a spectrum corresponding to 5,10,15,20-tetrafluorenoneporphyrin complex of zinc. This new porphyrin complex (4) was characterized by NMR, UV–visible, IR, microanalysis and mass spectrometry.
1H NMR (py-d5 in CDCl3): 8.98 (s, 8H, pyrrole), 8.51 (s, 4H, HD′), 8.35 (d, 3JHH = 7.2 Hz, 4H, HB′), 7.92 (d, 3JHH = 7.6 Hz, 4H, HA′), 7.84 (d, 3JHH = 6.4 Hz, 4H, HA), 7.82 (d, 3JHH = 6.4 Hz, 4H, HD), 7.67 (t, 3JHH = 7.2 Hz, 4H, HB), 7.45 (t, 3JHH = 7.3 Hz, 4H, HC. UV–visible (in CH2Cl2): 260 nm (fluorenone, ε260 = 370 714), 431 nm (Soret band, ε431 = 458 357), 556 nm (Q band, ε556 = 35 910), 599 nm (Q band, ε599 = 17 857). IR: 1715 cm−1 (CO band), 1616, 1455, 1180, 1105, 877, 736 cm−1. Analysis: calcd for C72H36N4O4Zn·4CH3OH C: 75.15, H: 4.32, N: 4.61, found C: 74.94, H: 4.33, N: 4.29 MS (ESI in CH2Cl2/CH3CN/pyridine): calcd for C72H36N4O4Zn: 1086.2033 [MH]+, found: 1086.2069 [MH]+.
By IR spectroscopy (KBr pellets), the νCO value observed for H2TFOP, [ZnTFOP] and [ZnTFOP.py] are unchanged at 1715 cm−1.
Acknowledgements
The authors are grateful to S. Sinbandhit (CRMPO), M. Werts (UMR CNRS 6510) and P. Le Maux for their technical assistance and helpful discussions.