

1 Introduction
Schiff bases constitute an interesting class of chelating agents. Due to their preparative accessibility, diversity and structure variability, they are capable of coordinating metal ions, giving complexes which exhibit wide applications in biological systems and industrial uses [1–3].
Schiff bases derived from ortho-phenylenediamine are of particular interest because of the proximity of the nitrogen atoms, which permits their simultaneous coordination to the same metal cation, leading mainly to stable monomer species [4–6].
However, these Schiff bases can be reduced to the corresponding amine derivatives, which have a higher stability because they are more flexible and not constrained to remaining planar [7–9], and they do not suffer the hydrolytic processes encountered with Schiff base analogues [10].
Our interest in the chemistry of reduced Schiff bases has also stemmed from previous results about antitumor activities of complexes of Cu(II) with a reduced Schiff base [11–13].
In the present work, we report the determination by potentiometric method of the protonation constants of a series of reduced Schiff bases (Fig. 1) derived from ortho-phenylenediamine, and stability constants of their complexes with Mn(II), Co(II), Ni(II) and Cu(II) ions.
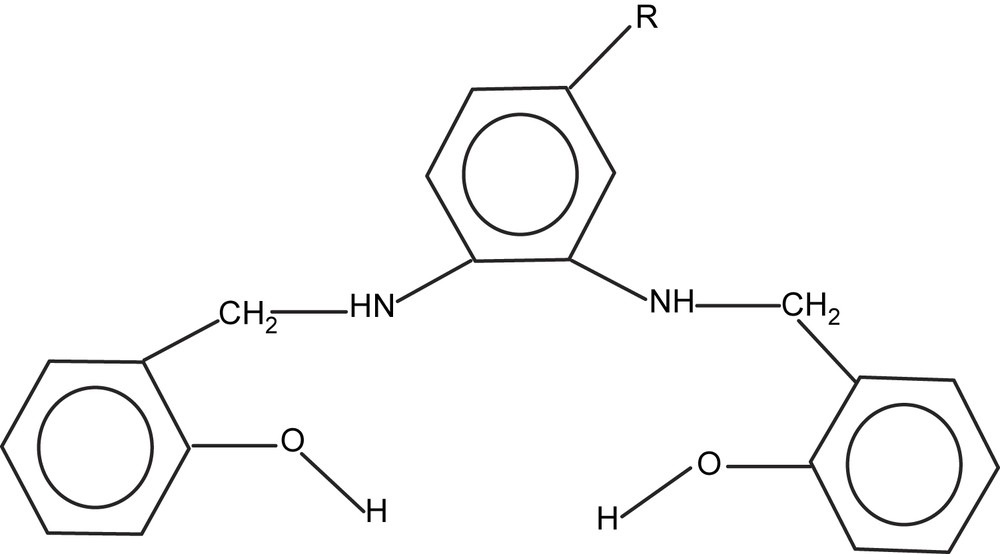
Formula of ligands: H2L1: R = H; H2L2: R = CH3; H2L3: R = Cl.
The aim of this study is the determination of both the effect of the R group and the reduction of the CN bonds on the stability of the complexes, by comparison of the results with those reported for Schiff base analogues.
2 Experimental
2.1 Reagents
ortho-Phenylenediamine, 3,4-diaminotoluene, 4-chloro-1,2-phenylenediamine and salicylaldehyde were purchased from Fluka and used without further purification.
Hexahydrated metal chlorides were obtained from Merck, exact concentrations of metal solutions were determined by titration with EDTA.
Carbonate-free sodium hydroxide used for the potentiometric titrations was prepared and standardized against a standard hydrochloric acid solution.
2.2 Potentiometric measurements
The potentiometric titrations were carried out in water–ethanol (90:10 v/v) solution at a 0.2 mol l−1 ionic strength (NaCl) in a thermostated vessel at 25.0 ± 0.1 °C under nitrogen atmosphere. The solutions were titrated with carbonate-free NaOH (0.1 mol l−1), the pH values were measured with a Hanna instruments HI 8417 pH meter.
The pH meter readings in water–ethanol (90:10 v/v) mixture pH(r) differs by an amount from the corrected reading in aqueous medium pH∗, according to the equation: pH∗ = pH(r) − δ [14]. The value of δ for water–ethanol (90:10 v/v) mixture was reported to be 0.02. Accordingly, the presence of 10% of ethanol in the solvent has a minor influence on the protonation or the complexes' stability constants obtained.
The measurements were carried out for the ligand alone at the concentration CL = 5 × 10−3 mol l−1, and for the ligand in the presence of metal cation (ligand/metal ratio 1:1), at the concentration CM = CL = 5 × 10−4 mol l−1.
The Sirko program [15] was used to calculate both protonation and stability constants of the ligands and their complexes. Computer-refined values of the constants, corresponding to the minimum standard deviation were retained, and used for the calculation of species distribution.
2.3 Synthesis of ligands
The ligands were synthesized by reduction of Schiff base analogues by NaBH4 [16]. The Schiff bases have been prepared by refluxing in ethanol 2 mmol of salicylaldehyde and 1 mmol of ortho-phenylenediamine for H2L1, 3,4-diaminotoluene for H2L2, 4-chloro-1,2-penylenediamine for H2L3. The resulting Schiff bases were recrystallized in ethanol, and then characterized by elemental analysis, NMR and IR.
In order to reduce the imine bonds, 1 mmol of Schiff base was dissolved in absolute ethanol, and 3 mmol of NaBH4 was added in small portions over 1 h. The resulting mixture was stirred for 2 h, during which time it changed color. Then, the solution was diluted and acidified with hydrochloric acid in order to destroy the excess of NaBH4.
The new ligand was extracted with CH2Cl2, the solvent was removed under vacuum and the resulting residue was washed with diethyl ether and stirred in hexane until it was transformed into a solid powder.
The elemental microanalyses were carried out at the Central Service of Analysis, CNRS Vernaison (France). The IR Spectra were recorded on a PerkinElmer spectrum one Fourier transform infrared spectrometer. 1H NMR was recorded on a Jeol Gsx 270 MSB (270 MHz) spectrometer.
3 Results and discussion
3.1 Characterization of ligands
Analytical data: found (calc.) for H2L1: C, 75.32 (75.00); H, 6.49 (6.25); N, 8.28 (8.75). For H2L2: C, 75.56 (75.44); H, 6.35 (6.58); N, 8.42 (8.38). For H2L3: C, 67.56 (67.70); H, 5.49 (5.35); N, 7.45 (7.89); Cl, 10.25 (10.01).
The analysis of IR spectra of the ligands shows the absence of any strong absorption at 1615 cm−1 attributable to the imine bond, indicating the reduction of CN bond to the C–NH one. This is confirmed by the presence of new absorption bands at 3390 cm−1 assigned to ν (N–H) of the amino groups [17].
The IR spectra of the ligands exhibit broad medium intensity band in the 3200–3270 cm−1 range, which is assigned to the O–H vibrations.
The aromatic rings give an intense band due to C–H deformation at 700 cm−1.
The 1H NMR spectra of the ligands (Table 1) have no azomethine hydrogen peaks at 8.2–8.5 ppm, but show new peaks at 4.2 ppm attributable to the methylene groups. The amine hydrogen peak was not observed.
1H NMR spectral chemical shifts (δ in ppm)
| Ligand | Aromatic protons | CH2–N | CH3 | –OH |
| H2L1 | 6.25–6.46 (m) | 4.30 (s) | 11.25 | |
| 6.72–7.02 (m) | ||||
| 6.89–7.08 (m) | ||||
| H2L2 | 6.05–6.18 (m) | 4.30 (s) | 2.42 (s) | 11.08 |
| 6.42–7.05 (m) | ||||
| 6.45–7.02 (m) | ||||
| H2L3 | 6.20–6.48 (m) | 4.30 (s) | 11.10 | |
| 6.58–7.12 (m) | ||||
| 6.45–7.06 (m) |
Nevertheless, these observations support the infrared data, and confirm the reduction of imine bonds to amine ones.
3.2 Potentiometric measurements
The constant βpqr calculated by the Sirko program are defined by the equilibrium:
| pM2+ + qH+ + rL2− ⇄ [MpHqLr](2p+q−2r)βpqr |
The successive constants log K for the ligands calculated from the overall constants log βpqr are listed in Table 2.
Protonation constants of the ligands (log K)
| Equilibrium | Log K | ||
| H2L1 | H2L2 | H2L3 | |
| L2−/HL− | 11.30 (±0.04) | 12.32 (±0.03) | 10.88 (±0.08) |
| HL−/H2L | 10.29 (±0.04) | 10.76 (±0.02) | 9.86 (±0.06) |
| H2L/H3L+ | 5.12 (±0.01) | 6.02 (±0.04) | 4.30 (±0.07) |
| H2L/H3L+ | 5.82 (±0.01) | 6.75 (±0.04) | 4.96 (±0.07) |
The ligands are tetradentate with two adjacent weak amine donors and two strongly basic phenolate groups. Four constants were obtained for each ligand; the basicity sequence obtained is H2L2 > H2L1 > H2L3.
The higher basic character of H2L2 is attributable to the electron-donor effect of the methyl group; and the lower basic character of H2L3 is due to the electron-withdrawing effect of the chloride atom.
Comparison of these results with those reported for Schiff base analogues [4–6] indicates that the protonation constants of these ligands are higher, especially those assigned to the nitrogen atoms of the amino groups. This arises because the nitrogen atoms of the Schiff base are not able to disperse charge to the solvent by hydrogen bonding [18].
The species distribution diagram as function of pH (Fig. 2) shows that the deprotonation of H4L2+ occur at pH = 4 and leads to the successive formation of H3L+, H2L, HL− and L2−, the deprotonation of both phenolic groups is completed at pH > 10.
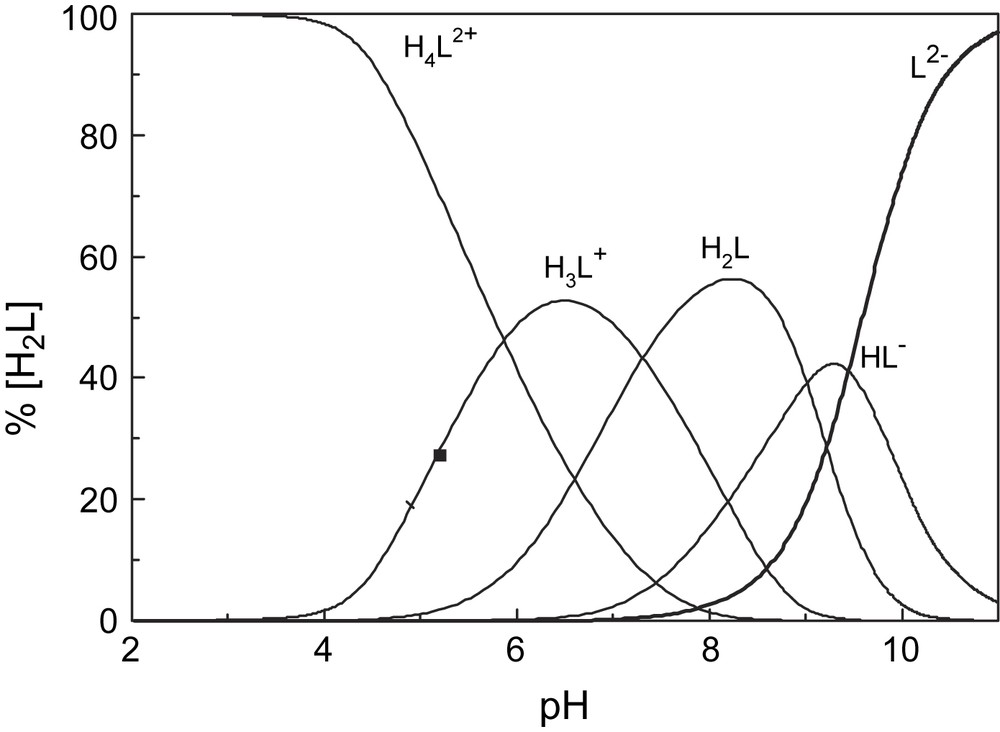
Species distribution diagram of H2L1.
Similar diagrams were obtained for H2L2 and H2L3.
The stability constants (log K) obtained for the complexes are listed in Table 3. For all the systems studied, only monomer species were formed, the model that best fits the experimental data is the one with formation of two species [MHL]+ and [ML].
Stability constants (log K) for the complexes
| Species | Mn | Co | Ni | Cu |
| [M(HL1)]+ | 7.94 (±0.01) | 10.32 (±0.10) | 10.58 (±0.08) | 10.95 (±0.05) |
| [ML1] | 14.23 (±0.04) | 16.41 (±0.06) | 16.85 (±0.04) | 17.31 (±0.01) |
| [M(HL2)]+ | 8.02 (±0.05) | 10.87 (±0.04) | 11.05 (±0.10) | 11.45 (±0.06) |
| [ML2] | 14.81 (±0.06) | 17.2 (±0.02) | 17.32 (±0.08) | 17.81 (±0.04) |
| [M(HL3)]+ | 6.13 (±0.01) | 9.81 (±0.07) | 9.74 (±0.09) | 10.21 (±0.07) |
| [ML3] | 12.48 (±0.07) | 15.31 (±0.05) | 15.22 (±0.03) | 16.1 (±0.08) |
The [MHL]+ species may be assumed to be protonated at the phenol oxygen of salicylaldehyde moiety. The complex [ML] results from the coordination of the metal to the ligand fully deprotonated. It is more stable than the complex [MHL]+, indicating that in the ML complex, the ligand is coordinated to the metal by both amino groups and phenolate oxygen atoms.
The systems H2L2–M(II) (M(II) = Ni, Cu, Co and Mn) give complexes that are more stable than those for the systems H2L1–M(II) due to the electron-donor effect of the methyl group, which is favorable to coordination.
For the systems H2L3–M(II), the opposite effect applies and the stability of their complexes are less than those for H2L1–M(II) systems, as a consequence of the electron-withdrawing effect of the chloride atom.
Thus, for any metal the stability sequence is H2L2 > H2L1 > H2L3 in agreement with the basic character order of these ligands.
For the systems ML (H2L = H2L1, H2L2, H2L3), the stability sequence observed is Cu > Ni > Co > Mn, in agreement with Irving–Williams series.
The complexes of Ni(II) and Co(II) with these ligands are more stable than those reported with Schiff base analogues [4–6]. In fact, the complexes of reduced Schiff bases are expected to be more flexible as they are not constrained to be planar.
The species distribution diagram for Ni–H2L3and Cu–H2L2 systems are reported in Figs. 3 and 4. Similar diagrams were obtained for the other systems.
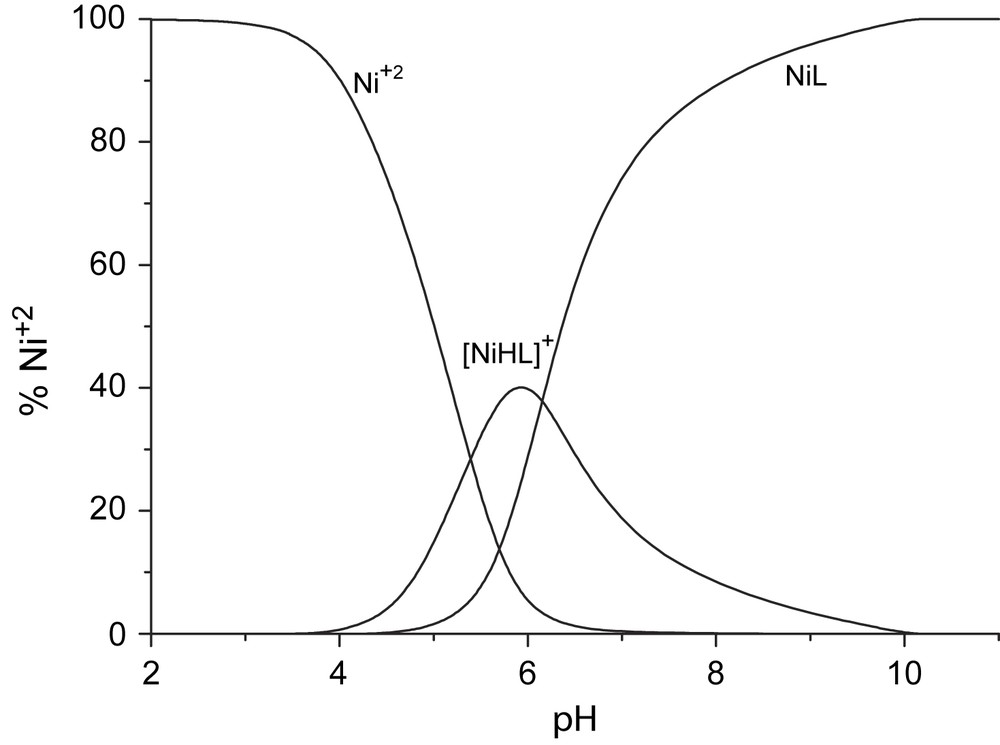
Species distribution diagram of Ni–H2L3.
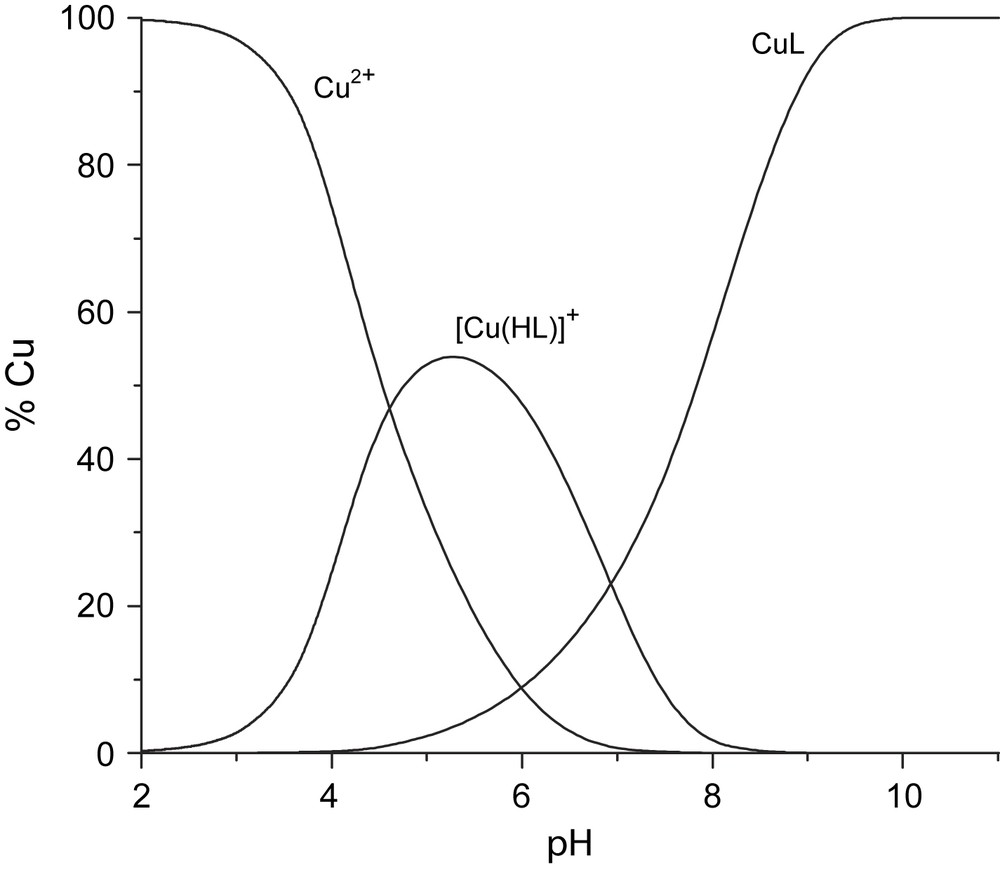
Species distribution Ni–H2L3 diagram of Cu–H2L2.