

1 Introduction
By virtue of its non-singlet spin state, oxygen, in the form of 3O2, is kinetically inert despite being an energetically very potent oxidant. Its oxidizing power is realized through the formation of ‘active oxygen’ species, i.e. singlet oxygen (1O2) and its reducing equivalents, the superoxide radical anion (O2−), hydrogen peroxide (H2O2) and hydroxyl radical (OH). Handling active oxygen is a major challenge both in vivo [1a] and in oxidation catalysis [1b], a challenge which frequently employs catalytic mediators to control the expression of their oxidizing power.
In vivo, the ability to deal with all forms of active oxygen is central to cellular protection. Indeed any imbalance in the decomposition of active oxygen species leads to oxidative cellular stress and, ultimately, loss of cellular viability. The mechanisms by which nature diffuses active oxygen safely involve the disproportionation of the active species to the inert oxygen species 3O2 and H2O, through a range of superoxide dismutases and hydrogen peroxide catalase enzymes or through reduction to H2O with ascorbate or glutathione [2]. Several of these catalase enzymes utilize dinuclear manganese active sites to effect these reactions, a key feature of which is the presence of carboxylate groups, which form bridges between the manganese centres [3,4]. Furthermore, manganese plays a key role in several other important biological processes and other manganese-containing enzymes include the oxygen evolving complex of photosystem II (PS II) [5,6], manganese superoxide dismutase [3,7] and arginase [8]. In this review, the nature and role of carboxylate bridging ligands in several of these enzymes, including dinuclear manganese based catalases [9,10] and arginases, and in structural and functional mimics will be explored. The role of carboxylate bridging ligands both in maintaining the dinuclear nature and in controlling the redox and hence ligand exchange chemistry of the complexes and enzyme active sites is discussed. The systems will be compared to related dinuclear manganese complexes employed in the catalytic oxidation of organic substrates, with regard to mechanistic aspects of the catalysis.
2 Dinuclear manganese catalase enzymes
Although the majority of known catalase enzymes are iron-heme based enzymes, several organisms utilize dinuclear manganese-containing enzymes to disproportionate hydrogen peroxide [9,10]. Two crystal structures at atomic resolution have been obtained for enzymes isolated from Thermus thermophilus (TTC) [11,12] and Lactobacillus plantarum [13]. The enzymes originating from both species consist of six identical subunits, each containing a manganese dimer in the active site. In T. thermophilus [12] two conformations (forms I and II) of the enzyme can be distinguished. The manganese ions are bridged by a μ-carboxylate ligand (Glu70) and μ-oxygen bridges (either aqua, hydroxo or oxo ligands). The Mn ions are each coordinated to one His residue and one Glu residue. For Mn(2) the Glu is bound via one oxygen, resulting in penta-coordination, while Mn(1) is coordinated to a (labile) terminal molecule of water also, rendering the Mn ion hexa-coordinate. The crystal structure for the dinuclear manganese catalase enzyme from L. plantarum [13] reveals a similar 1st coordination sphere (Fig. 1). The manganese ions are bridged by a μ-carboxylate from Glu66 and contain two more oxygen bridges, most probably one μ-oxo and one μ-hydroxo (in the resting, Mn2III, state). Furthermore, one of the Mn ions is bound to one His181 and a chelating Glu148 carboxylate, while the second Mn ion is bound to one His69 and a monodentate Glu35 carboxylate, with the sixth coordination site again being occupied by a (labile) water molecule.
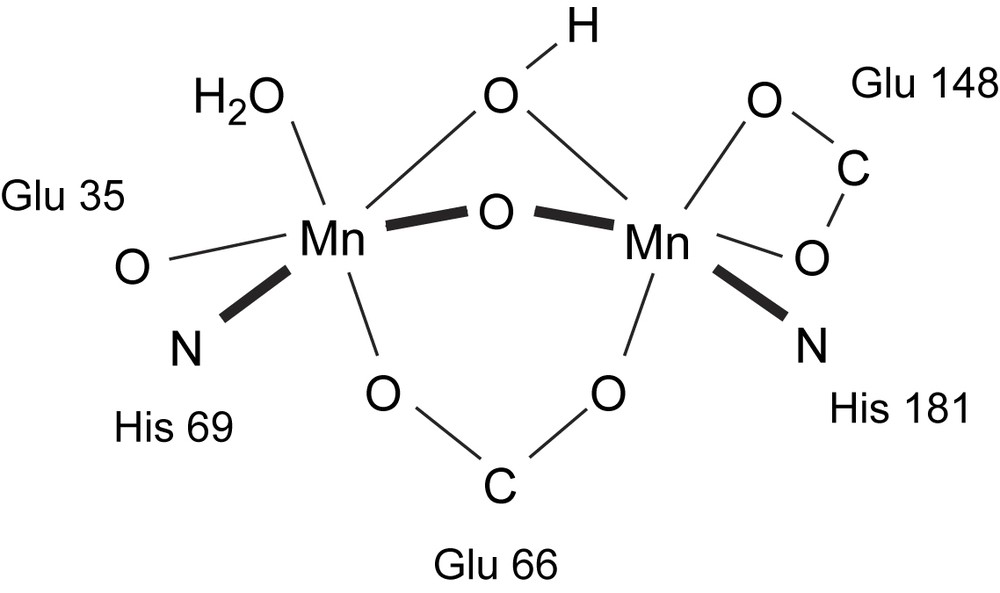
The active site of L. plantarum catalase [13].
Four oxidation states are accessible in the dinuclear manganese catalase enzymes (Mn2II, MnIIMnIII, Mn2III and MnIIIMnIV) [14,15]. The majority of the as-isolated enzyme was in the Mn2III state, although residual Mn2II species and the inactive superoxidized MnIIIMnIV state (vide infra) were also present [16,17].
The activity of these enzymes is dependent on both the pH [10,18] and the oxidation state of the dinuclear manganese centre. During the catalytic decomposition of H2O2 the enzyme cycles between the Mn2III and Mn2II states [14,15] and shows similar activity when starting either in the Mn2II and Mn2III redox states [15,20]. Treatment with NH2OH [16,19,69], which serves either as one or as two-electron reductant, depending on local pH [14], in the presence of H2O2 leads to complete conversion of the enzyme to the inactive MnIIIMnIV state (via MnIIMnIII). In contrast, reduction with NH2OH in the absence of H2O2 of either the Mn2III enzyme or the inactive MnIIIMnIV superoxidized form yields the Mn2II state, with full restoration of activity [16]. The interconversion between the different redox states is summarized in Fig. 2.
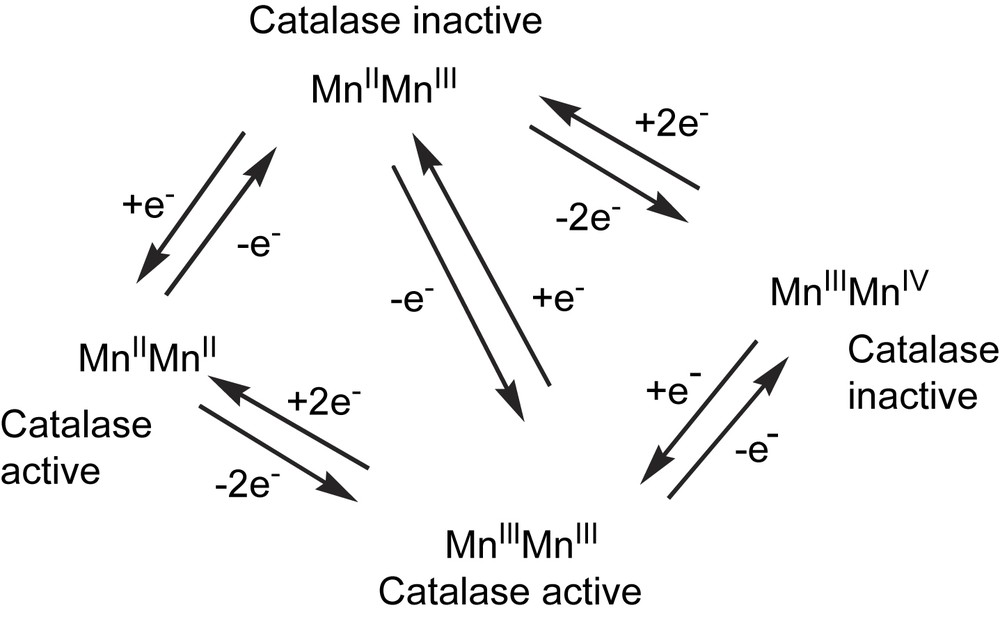
Interconversion between the different redox states of the manganese catalase enzymes. H2O2 acts both as a two-electron oxidant and reductant, whereas NH2OH acts, predominantly, as a one-electron reductant [16,69].
When the enzyme is in the Mn2II state it can be inhibited by the (reversible) binding of several anions (e.g. Cl−, F−, HPO4−) [11,20] and consequently, when H2O2 decomposition is performed in the presence of either Cl− or F− the enzyme is trapped in Mn2II state [15]. Inhibition by chloride has been confirmed by X-ray crystallography to be due to the replacement of a bridging μ-oxygen ligand by a bridging μ-Cl− ligand [11,12]. In contrast, azide binds to the terminal position of Mn(1) (in the Mn2III state, Fig. 1) by displacing the terminal labile water molecule from the native enzyme in a mode, which is, possibly, similar to the (initial) binding of H2O2 [13].
As mentioned above, the active site of manganese catalase enzymes contains three distinct solvent molecules. From X-ray crystallographic analysis (on the native Mn2III enzyme), two solvent molecules bridge between the two Mn centres, while the third water molecule occupies a terminal position on one of the Mn ions. The lability of both the terminal water and the oxygen bridges (either being μ-aqua, μ-hydroxo or μ-oxo) is central to the catalytic activity as exemplified by both Cl− and N3− inhibition (vide supra). This lability is dependent on the oxidation state of the manganese centres, the Mn–Mn separation and protonation state of the oxygen bridges [10,13,21]. For the Mn2III state, it has been proposed that the solvent bridges are μ-hydroxo (trans-His) and μ-oxo (trans-Glu), while for the Mn2II state the solvent bridges are believed both to be singly protonated, i.e. both are μ-hydroxo [12,13]. The protonation of the μ-oxo bridges increases their lability and may play a role in governing the differences in the interaction of H2O2 with the Mn2II and Mn2III states [13,14]. Based upon magnetization studies (on TTC) two different pH-dependent bridging modes {(Mn2III(μ-O)(OH2)(OH) (open) and Mn2III(μ-O)2 (closed)} for the Mn2III state have been proposed (Fig. 3) [22].
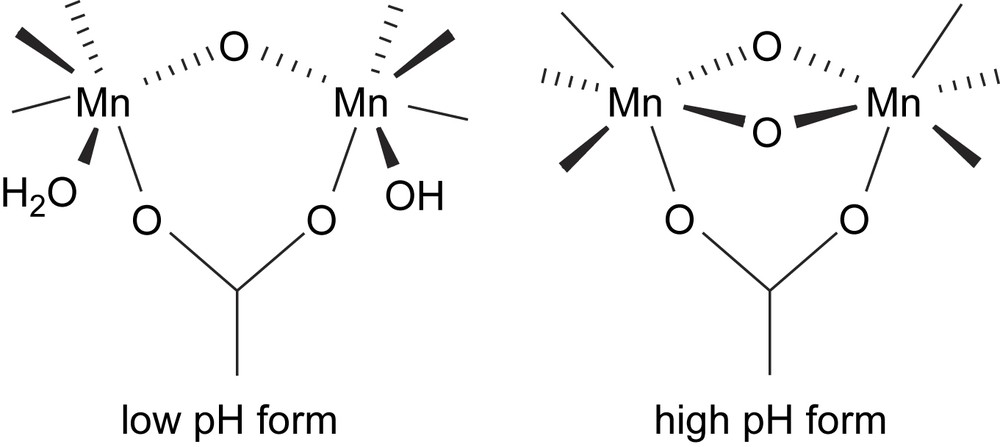
Proposed pH-dependent equilibrium between open and closed Mn2III catalase enzyme active site [22].
During catalytic turnover the dinuclear manganese catalase enzymes cycle between Mn2III and Mn2II and one molecule of H2O2 is oxidized to O2, while another molecule is being reduced to H2O (Fig. 4) [10,11,14]. For the oxidative and reductive half-reactions several different coordination modes for H2O2 have been proposed. During the oxidative half-reaction H2O2 replaces the terminal H2O ligand on one of the Mn centres in the Mn2III complex, and protonates the μ-oxo bridge.1 Subsequent reduction of the Mn-dimer results in the formation and release of O2. The second equivalent of H2O2 binds to the Mn2II as a bridging μ1,1-hydroperoxo (either associatively via initial terminal coordination weakening the bridging water, or dissociatively via initial loss of labile H2O). The μ1,1-bridging mode polarizes the O–O bond and may even be polarized further by gem-protonation of hydrogen peroxide, thus facilitating heterolytic O–O bond cleavage, closing the catalytic cycle by reoxidation to Mn2III with loss of water.
It is worth noting that, in addition to the manganese catalase enzymes, several other manganese-containing enzymes have been tested for catalase activity and, although their biological role is different from that of catalases, they are nevertheless active [26]. An example is arginase, whose physiological role is to cleave l-arginine to l-ornithine hydrolytically and thus plays an important role in mammalian nitrogen metabolism [3,23]. The active site consists of a Mn2II dimer (which does not undergo redox change during the hydrolytic catalysis), bridged by two aspartates and one μ-H2O (Fig. 5). It has been proposed recently that upon binding of arginine to one of the Mn centres, the μ-H2O bridge is opened. Deprotonation of the H2O ligand provides a nucleophilic hydroxide, which then attacks the substrate [24].
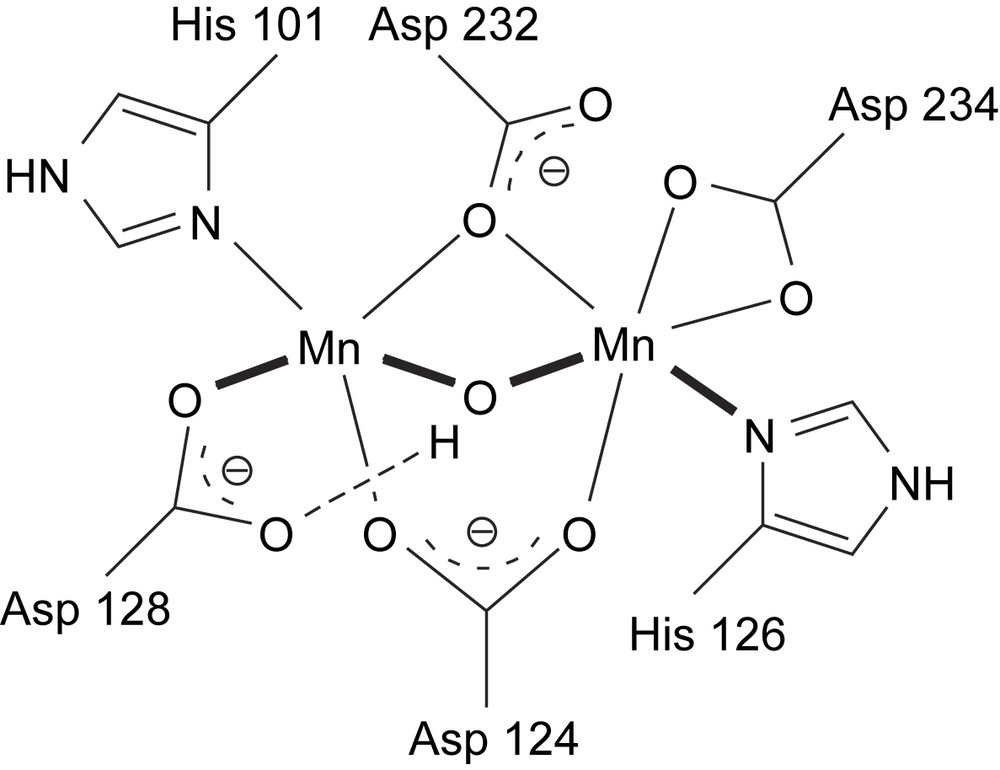
Active site of arginase enzyme containing a Mn2II dimer [25,26].
The catalase activity (kcat/KM) of arginase is five orders of magnitude lower than the catalase enzymes, however, its ability to exhibit catalase activity demonstrates the importance of the dinuclear manganese core [26]. The reason for the lower catalase activity of arginase may be due either to the lack of a suitable proton donor/acceptor near the active site (e.g. Lys162 in TTC), which facilitates proton-transfer or the difference in the third bridging ligand (for arginase a μ1,1-carboxylate bridge and for manganese catalases a μ-oxo bridge) in stabilizing the Mn2II redox state or both.
Overall it is apparent that the dinuclear μ-carboxylate bridge structural motif is key to the activity of these enzymes in addition to the availability of labile coordination site(s). Importantly, the catalytic activity of these enzymes does not involve loss or partial dissociation of the carboxylate bridge, although in other dinuclear manganese or iron enzymes a carboxylate shift has been found to play an important role in enzyme function (e.g. MMO) [27,28].
3 Structural, functional and spectroscopic models for catalase enzymes
Modeling the active site of dinuclear manganese catalase enzymes both in terms of structure and function has, over the last two decades, focused on the use of multidentate ligand systems, most notably in the systems described by the groups of Dismukes [29,30], Wieghardt [31,32], Pecoraro [33], Sakiyama [34–37] and others [38]. In the present review we will focus on the functional and structural models for the catalase enzymes, which present similar dinuclear carboxylate-bridged cores2 [39]. The majority of systems reported are based on a relatively few, albeit structurally diverse, set of non-bridging ligands, in particular phenols [34,37,40], polypyridyl ligands [41–43], tris(pyrazoyl)-borates [44] and tris(imidazoyl)-phosphines [45]. Overall, four remarkable aspects regarding the diversity in the ligand systems employed in modeling catalase enzymes are (i) that structurally very similar complexes can exhibit remarkably different behaviours with respect to both catalase activity and in oxidation catalysis, (ii) that the lability of ligands, in particular water, is central to activity and decreases with increasing oxidation state of the manganese ions, (iii) that whereas in enzymatic systems the carboxylate bridging ligand is generally accepted to be stable and oxo, aquo and aqua ligands to be labile with respect to dissociation during catalysis, in biomimetic studies (vide infra) the reverse is frequently perceived to be the case, and (iv) while μ-oxygen bridges generally facilitate communication between two Mn centres, carboxylate bridges both increase the Mn–Mn separation and electronically shield the Mn centres, thus promoting two-electron processes instead of two subsequent one-electron processes to occur3 (e.g. Mn2II/Mn2III instead of Mn2II/MnIIMnIII/Mn2III).
3.1 TACN-based structural models
A structurally and electronically diverse series of mono-, di-, tri- and tetranuclear manganese complexes can be obtained with both the tacn (1,4,7-triazacyclononane) and tmtacn (N,N′,N″-trimethyl-1,4,7-triazacyclononane) family of ligands (Fig. 6) [46–52]. For the dinuclear complexes a series of redox states (Mn2II to Mn2IV) are accessible containing μ-carboxylate bridges and/or μ-oxygen bridges. The solid-state (i.e. X-ray crystallography) and solution chemistry of these different dinuclear manganese (carboxylate) complexes have been examined in detail and demonstrate the propensity for dinuclear manganese systems to undergo rearrangement of their bridging ligands in response to changes in redox state [53–58]. Indeed, the lower oxidation states of the complexes described favour carboxylate and aquo bridging ligands whereas the higher oxidation states favour μ-oxo bridging ligands.
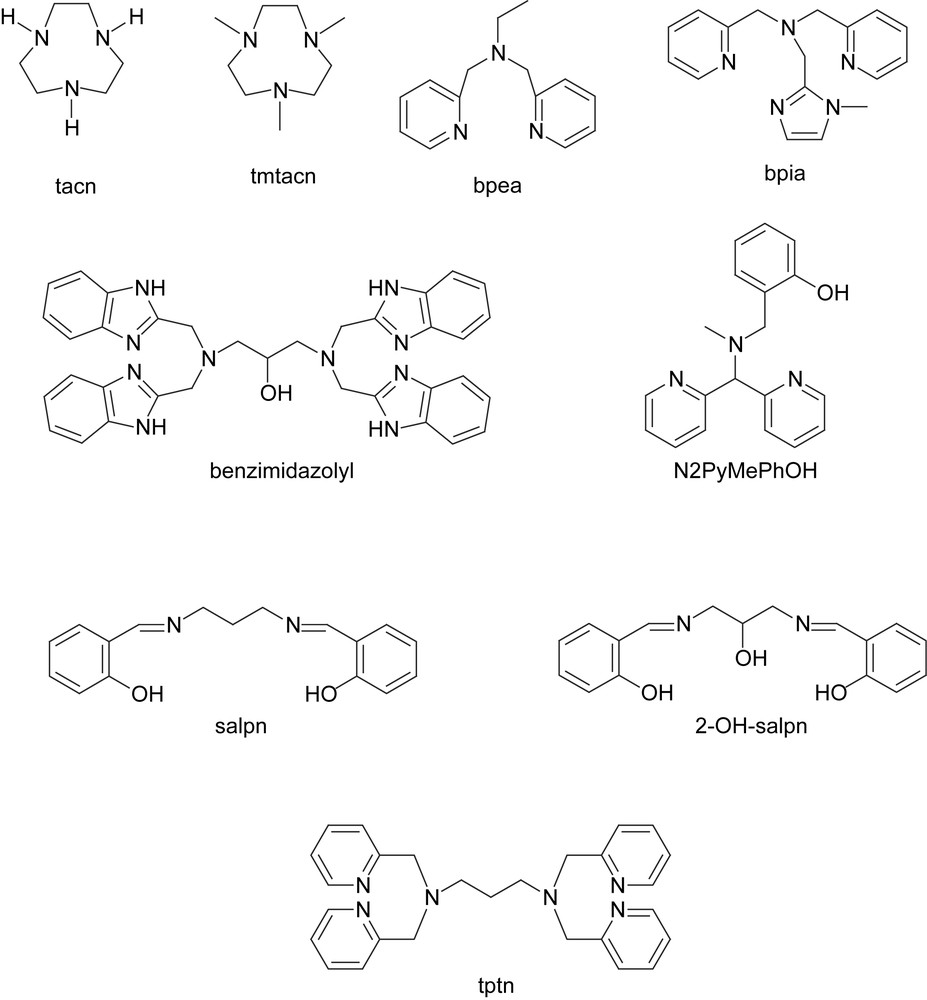
Ligands discussed in the text.
Bis-carboxylate [Mn2III(μ-O)(μ-CH3CO2)2L2]2+ complexes (with L = tacn or tmtacn) exhibit dynamic solution chemistry [31]. For example, [Mn2III(μ-O)(μ-CH3CO2)2(tmtacn)2]2+ can exhibit the Mn2IV/MnIIIMnIV/Mn2III/MnIIMnIII redox couples in (anhydrous) acetonitrile, retaining the mono-μ-oxo/di-μ-carboxylate core. The same holds for [Mn2II(μ-OH)(μ-CH3CO2)2(tmtacn)2]+, which can be oxidized by two separate, reversible one-electron processes: Mn2III/MnIIMnIII/Mn2II. On the other hand, in aqueous solutions disproportionation reactions can occur resulting in the formation of for example [Mn4IV(μ-O)6(tacn)4]4+ and [Mn2IV(μ-O)3(tmtacn)2]2+ [46,31].
The latter complex [Mn2IV(μ-O)3(tmtacn)2]2+, although kinetically stable towards ligand exchange, can undergo electrochemically induced ligand exchange reactions of the μ-oxo bridges in carboxylate containing buffer systems, forming carboxylate-bridged [Mn2III(O)(μ-RCO2)2(tmtacn)2] complexes [59]. Mixing equimolar amounts of [Mn2III(μ-O)(μ-CH3CO2)2(tmtacn)2]2+ and [Mn2III(μ-O)(μ-CH3CO2)2(tacn)2]2+ results in slow formation of mixed ligand species [Mn2III(μ-O)(μ-CH3CO2)2(tmtacn)(tacn)]2+ [60] as shown by 1H NMR spectroscopy [61]. The lability of both the μ-oxo and μ-acetate bridges is demonstrated further by the formation of mononuclear complexes of the type [MnIII(X)3(tmtacn)] (X = N3−, Cl− or NCS−) when [Mn2III(μ-O)(CH3CO2)(tmtacn)2]2+ is reacted with the corresponding anion in ethanol [31,62].
[Mn2III(μ-O)(μ-CH3CO2)2(tacn)2]2+, under aerobic conditions in acidified aqueous media, forms the mixed-valent [Mn2III,IV(μ-O)2(μ-CH3CO2)(tacn)2]2+ complex (in which one of the acetates is replaced by μ-oxo bridge). By contrast [Mn2III(μ-O)(μ-CH3CO2)2(tmtacn)2]2+ retains both acetate bridges to form [Mn2III,IV(μ-O)(μ-CH3CO2)2(tmtacn)2]3+ in (anaerobic) acidic aqueous solution [31,63]. Subsequent replacement of the μ-acetate in [Mn2III,IV(μ-O)2(μ-CH3CO2)(tacn)2]2+ by either two chloride or fluoride ligands yields [Mn2IV(μ-O)2(tacn)2(X)2]2+ (X = F−, Cl−) with a terminal bound halide anion on each Mn centre upon (aerobic) reaction in water with either NaBF4 or conc. HCl, respectively [63].
Surprisingly, despite their structural diversity the catalase activity of this family of complexes has received relatively little attention, in part due to its remarkable catalytic activity with respect to oxidation of organic compounds with hydrogen peroxide (vide infra). Indeed, considerable effort has been expended on suppressing catalase activity.4 Nevertheless a Mn-tmtacn dinuclear complex has been reported, the first to contain a μ-peroxo bridge. The complex [Mn2IV(O)2(μ-O2)(tmtacn)2] releases O2 at room temperature, yielding, initially, a Mn2III complex which undergoes rapid disproportionation to the MnII and Mn2IV states [64]. The observation of this bridging mode is of particular relevance to the oxidation of H2O2 during catalase activity.
Mixed ligand complexes based upon Mn-tmtacn were also prepared: both [(tmtacn)MnIV(μ-O)2(μ-CH3CO2)MnIII(CH3CO2)2] and [(tmtacn)MnIV(μ-O)2(μ-CH3CO2)MnIII(bipy)(MeOH)]2+ [32]. Both complexes showed catalase activity in aqueous acetate buffer and the activity was approximately five orders of magnitude lower than the natural enzymes.
3.2 Bpea-based catalase mimics
As for the tacn and tmtacn family of ligands, the bpea (N,N′-bis(2-pyridylmethyl)-ethylamine, Fig. 6) ligand allows the preparation of a diverse range of dinuclear manganese complexes in several oxidation states and bridging modes, including Mn2II(μ-CH3CO2)3, Mn2III(μ-O)(μ-CH3CO2)2, Mn2III,IV(μ-O)2(μ-CH3CO2) and Mn2IV(μ-O)2(μ-CH3CO2) cores which show electrochemically induced interconversion [65]. As for the catalase enzymes, the catalytic cycle for the catalase activity exhibited by these types of complexes is proposed to involve a Mn2II/Mn2III couple as depicted in Fig. 7, with ‘opening’ of the μ-oxo bridge of the Mn2III complex being central to allow ligand exchange with H2O2. For [(bpea)2Mn2II(μ-CH3CO2)3]+ catalase activity was found to decrease with increasing acetate concentration (the measured O2 evolution rate decreases from 1.8 to 0.9 to 0.5 ml/min in the presence of 0, 1 and 5 equiv. of NaOAc with respect to complex, respectively), assigned to the stabilization of the tris-acetate bridged complex in which the labile sites are ‘blocked’ to H2O2 coordination. However, it should be noted also that the addition of acetate leads to a change in pH, which favours the formation of the μ-oxo bridged Mn2III complex, and thereby may prevent ligand exchange with H2O2.
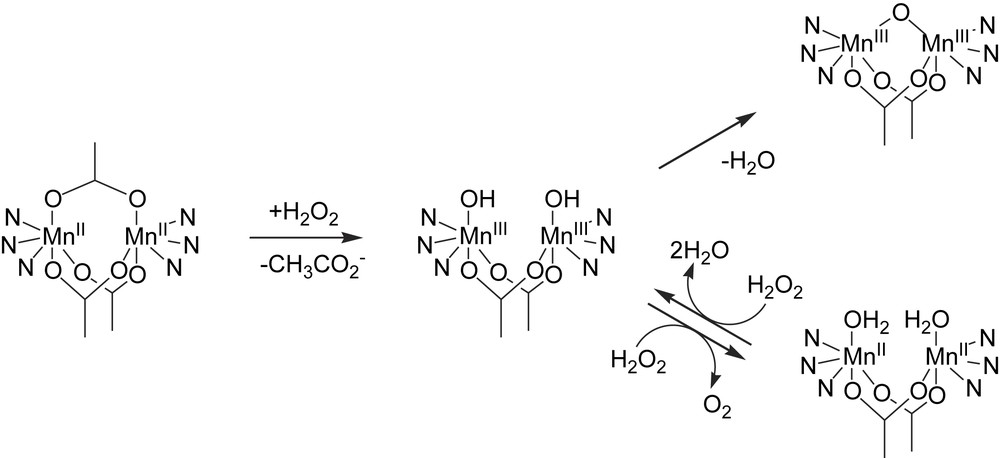
Catalase activity by [(bpea)2Mn2II(CH3CO2)3]2+ [65].
3.3 Bpia-based catalase mimics
A series of (mono- and dinuclear) manganese complexes based on the ligand bpia (bis(picolyl)(N-methylimidazol-2-yl)amine, Fig. 6) with various oxidation states and bridging modes have been reported by Krebs and Pecoraro [66], e.g. [Mn2II(μ-CH3CO2)2(bpia)2]2+, [Mn2III(μ-O)(μ-CH3CO2)(bpia)2]3+, [Mn2III,IV(μ-O)2(bpia)2]3+ and [Mn2III(μ-O)(bpia)(Cl)2]2+. Although H2O2 is an efficient reductant of the chloride complex, containing a single μ-oxo bridge and a terminal chloride on each Mn centre, it is incapable of reoxidising the reduced complex, and hence this complex does not exhibit catalase activity. By contrast, [Mn2II(μ-CH3CO2)2(bpia)2]2+ is a very efficient manganese catalase model with a kcat/KM of only two to three orders of magnitude lower than that of the manganese catalase enzymes. Similarly, [Mn2III(μ-O)(μ-CH3CO2)(bpia)2]3+ showed catalase activity and was converted to the [Mn2III,IV(μ-O)2(bpia)2]3+ complex in the presence of H2O2.5
3.4 Benzimidazolyl-based catalase mimics
In a series of reports, Dismukes and coworkers described the redox chemistry and catalase activity of a series of binuclear manganese complexes based on chelating heptadentate, benzimidazolyl based ligand (L) (N,N′,N″,N‴-tetrakis(2-methylenebenzimidazolyl)-1,3-diaminopropan-2-ol, Fig. 6) [67]. The complexes isolated initially were [Mn2II(L)(μ-Cl)(Cl)2] and [Mn2II(L)(μ-OH)(Br)2], containing a μ-chloride bridge and a μ-hydroxo bridge, respectively, in addition to a μ-alkoxy bridge from the ligand. The complexes were isolated in the Mn2II oxidation state, however, during H2O2 decomposition (typically up to 200 t.o.n.) the predominant oxidation state was found to be Mn2III(μ-O), determined by UV–vis and IR spectroscopy, suggesting that a Mn2II/Mn2III cycle was in operation, i.e. similar to that observed for the manganese catalase enzymes.
Three separate redox processes were observed for [Mn2II(L)(μ-OH)(Br)2] complex (i.e. Mn2II/MnIIMnIII/Mn2III/MnIIIMnIV), while for the μ-Cl bridged complex a one electron, followed by a two-electron oxidation was observed (Mn2II/MnIIMnIII/MnIIIMnIV) [67]. However, upon replacement of the halide by an acetate ligand, i.e. [Mn2II(L)(μ-CH3CO2)]2+ (Fig. 8), the first oxidation becomes a two-electron process followed by an one electron oxidation, i.e. Mn2II/Mn2III and Mn2III/MnIIIMnIV [68]. The effect of the acetate bridge in the series of complexes based upon L is intriguing. While for both the related μ-Cl and the μ-OH complexes a Mn2II/MnIIMnIII couple is observed (vide supra; E1/2 = 0.49 and 0.54 V vs. SCE respectively), the μ-acetate complex shows a two-electron Mn2II/Mn2III process at 0.81 V [68,69]. Thus introduction of a μ-acetate bridge induces a two-electron process in place of two sequential one electron processes, thus circumventing the mixed-valent MnIIMnIII state. The behavior of the [Mn2II(L)(μ-CH3CO2)] complex with water, hydroxide and oxygen (in acetone) was studied by several techniques (electrochemistry, 1H NMR, ESR, FT-IR and MS spectroscopies) [2]. The penta-coordinate dinuclear manganese complex [Mn2II(L)(μ-CH3CO2)]2+ shows a two-electron Mn2II/Mn2III process (in acetone), while addition of one equivalent of hydroxide changes this to two single electron processes (Mn2II/MnIIMnIII/Mn2III), attributed to the formation of hexa-coordinate [Mn2II(L)(μ-CH3CO2)(μ-OH)]+. Addition of another equivalent of hydroxide is proposed to yield [Mn2II(L)(μ-CH3CO2)(OH)2] with two terminal hydroxides, leading to the (re)appearance of a two-electron processes (Mn2II/Mn2III/Mn2IV). Overall, the redox behavior suggests that the acetate remains bound as a bridging ligand and the changes (from 2e− to 1e− processes, and vice versa) can be rationalized by the differing coordination modes of the oxygen ligands (i.e. hydroxide), with μ-O/μ-OH, μ-Cl and μ-Br ligands facilitating electronic communication between the manganese centres. By contrast the acetate bridges serve to reduce electronic communication and allow for two-electron redox processes to take place.
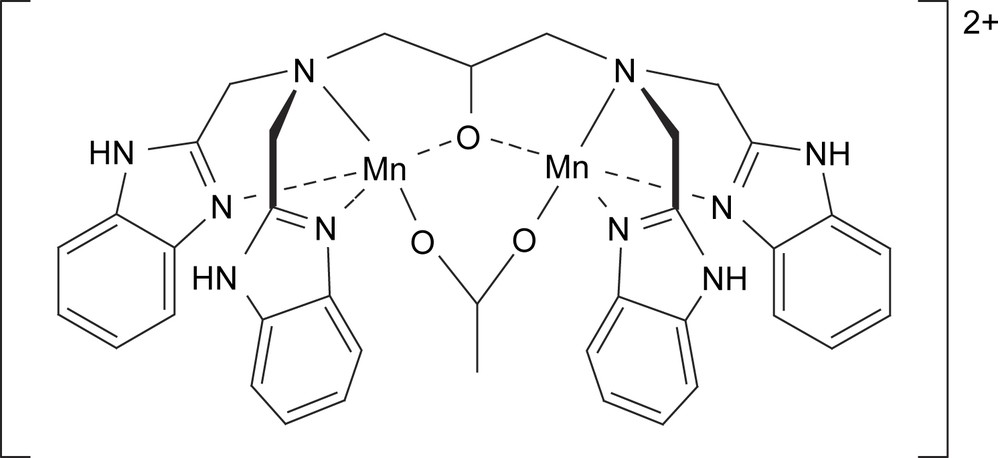
[Mn2II(CH3CO2)(L)]2+ complex as a functional model for catalase [68].
From UV–vis and EPR spectroscopies and kinetic studies the catalase activity of the acetate bridged complex was determined to involve a Mn2II/Mn2III cycle [29]. Two notable observations with respect to water and acetate content of the reaction mixture were made: i.e. the use of up to 2 v/v% of water (in MeOH) resulted in a decrease in the lag-time observed normally, while above 2 v/v% water,6 a lower catalase rate was observed. Addition of (tetra-n-ethylammonium) acetate resulted in a decrease in the rate of H2O2 decomposition (4-fold reduction in rate for 20 equiv. of acetate). These results were initially interpreted as shown in the following mechanistic scheme (Fig. 9) [29]. Acetate inhibition was thought to arise from coordination of a second μ-acetate bridge to the five-coordinate manganese centres, while replacement of the single μ-acetate bridge by two terminally bound water molecules was proposed to be responsible for the lag-time observed (while at higher water concentration binding of water competes with peroxide). Alternatively, the binding mode of the acetate might simply change from bridging to terminal monodentate.
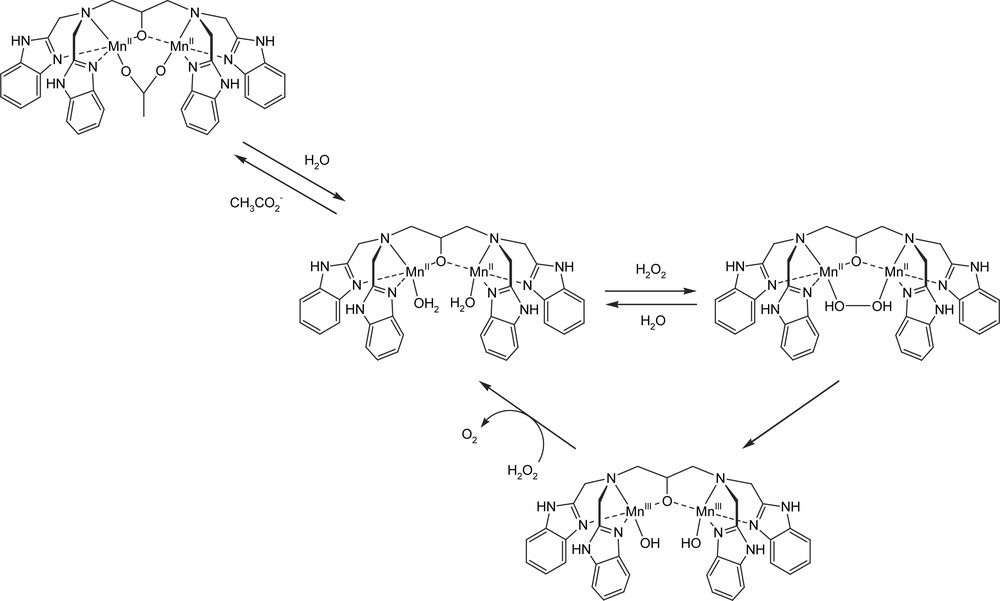
Proposed mechanistic scheme [29].
The kinetics of the catalytic decomposition of H2O2 by [Mn2II(L)(μ-CH3CO2)]2+ in MeOH and MeOH/H2O (98:2 and 11:89) was studied by monitoring the rate of O2-evolution in a more recent study. The species present during and after catalysis were characterized by UV–vis and 1H NMR spectroscopies and structural assignments made, primarily by comparison with species assigned in earlier studies [30] [Mn2II(L)(μ-CH3CO2)]2+ is an efficient catalase mimic, reaching > 2000 t.o.n. with no detectable catalyst decomposition. As for the related catalase enzyme, H2O2 disproportionation proceeds via a Mn2II/Mn2III cycle. The lag-phase normally observed for this reaction was shown to either decrease upon pre-equilibration with water (vide supra) or was completely eliminated upon pretreatment with one equivalent of hydroxide (and the steady state rate of H2O2 decomposition increases linearly with up to 5 equiv. of hydroxide). Addition of one equivalent of hydroxide converts [Mn2II(L)(μ-CH3CO2)]2+ to [Mn2II(L)(μ-CH3CO2)(μ-OH)]+ (based upon electrochemistry). While the effect of water was rationalized by the formation of equal amounts of the (active) [Mn2II(L)(μ-CH3CO2)(μ-OH)]+ species (via deprotonation of [Mn2II(L)(μ-CH3CO2)(μ-H2O)]2+ formed initially) and (inactive) protonated complex [Mn2II(LH)(μ-CH3CO2)(μ-H2O)]3+ (observed by ESR as an uncoupled Mn2II species). The catalase rate shows saturation kinetics at high H2O2 concentrations, indicating peroxide/catalyst complex formation.
Two species appeared to be important precursors (i.e. [Mn2II(L)(μ-CH3CO2)(μ-OH)]+ and [Mn2III(L)(μ-CH3CO2)(μ-OH)]3+) to the catalytically active species [2,30]. The reactivity of [Mn2II(L)(μ-CH3CO2)(μ-OH)]+ is increased by either addition of hydroxide or water (the non-coordinating base 2,6-di-tbutylpyridine has no effect) and upon reaction with H2O2 [Mn2III(L)(μ-CH3CO2)(μ-OH)]3+ undergoes a change to a species proposed to be [Mn2III(L)(CH3CO2)(OH)(μ-O)]+, in which the acetate bridge has changed its coordination mode to a terminal position (Fig. 10). During catalytic turnover, [Mn2II(L)(μ-CH3CO2)(μ-OH)]+ is proposed to be in equilibrium with ‘open’ species [Mn2II(L)(μ-OH)(CH3CO2)(H2O)]+ in which the acetate has shifted to a terminal position. The terminal water undergoes ligand exchange with H2O2 and the terminal peroxo ligand subsequently exchanges with the μ-OH to form a μ,η2-peroxo bridge. In the next step the O–O bond of the peroxide is cleaved and the complex undergoes two-electron oxidation to [Mn2III(L)(CH3CO2)(OH)(μ-O)]+. Binding of a second H2O2 yields a terminally bound peroxide and subsequently O2 is released together with reduction of the complex to reform [Mn2II(L)(μ-OH)(CH3CO2)(H2O)]+ and closing the catalytic cycle.
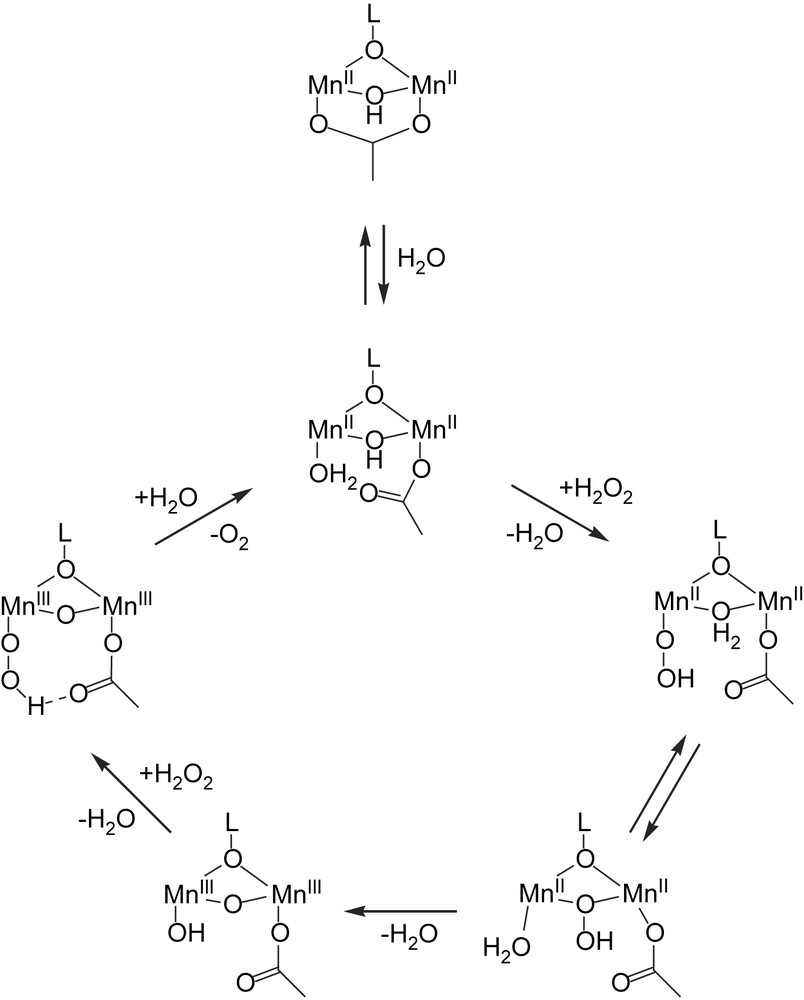
Proposed catalytic cycle for catalase activity of [Mn2II(L)(μ-CH3CO2)]2+ [30].
3.5 SALPN-based catalase mimics
Carboxylate shifts, although implicit in the conclusions of Dismukes and coworkers, are not suspected as to play a significant role in effecting H2O2 disproportionation in the catalase enzymes themselves. Indeed in the SALPN (1,3-bis(salicylideneamino)propane, Fig. 6) based series of catalase mimics, carboxylate bridging ligands are not present, yet they have shown catalase type activity also. The dinuclear manganese complexes based on SALPN ligands operate as binuclear catalysts through a Mn2II/Mn2III redox cycle, reminiscent of the redox cycle of the catalase enzymes7 [70–73]. The SALPN ligands react with manganese(III) acetate to form a large variety of complexes depending upon reaction conditions, including mono- and dinuclear complexes and polymeric chains [74,75]. Reaction of these ‘MnIII(SALPN)’ complexes with H2O2 yields dinuclear [Mn2IV(SALPN)(μ-O)2] complexes [76,77].
Modification of the SALPN ligand by incorporation of an alcohol functional group into the propane bridge, i.e. 2-OH-SALPN (1,3-bis(salicylideneamino)-2-propanol, Fig. 6), allows similar dinuclear complexes to be obtained. Dinuclear [Mn2III(2-OH-SALPN)2] acts as an efficient functional model for manganese catalase enzymes and the corresponding Na2[Mn2II(2-OH-SALPN)2]·2MeOH has been prepared under anaerobic conditions also [78]. The catalytic cycle for H2O2 decomposition is proposed to involve a Mn2II/Mn2III cycle and, as for the manganese catalase enzymes, the combination of H2NOH and H2O2 results in the formation of a catalytically inactive MnIIIMnIV dimer. Reactivity can be restored by reaction with H2NOH in the absence of H2O2.
Kinetic studies on a series of [Mn2II(2-OH-X-SALPN)2]2− (X = 5-OCH3, H, 5-Cl, 3,5-diCl, or 5-NO2) complexes with electron donating and electron withdrawing substituents showed that these systems are effective functional mimics of dinuclear manganese catalase enzymes [33,79]. The 2-OH-SALPN based dinuclear manganese complexes are bridged by two alkoxides and each of the manganese dimers is further coordinated to two oxygen and two nitrogen atoms from the 2-OH-SALPN ligands. As with catalase, the catalytic cycle involves the Mn2II/Mn2III couple. An inactive MnIIIMnIV is formed when oxygen is present at high concentration during H2O2 decomposition, however, reactivity can be restored upon reaction with H2NOH. The catalyst can undergo at least 5000 t.o.n. Upon binding of H2O2 to [Mn2III(2-OH-(X)-SALPN)2], one of the alkoxide bridges changes its coordination mode from bridging to monodentate (alkoxide shift), thus facilitating terminal coordination of the peroxide to the other Mn ion (Fig. 11). Subsequent reduction of the Mn-dimer and release of O2 is the rate limiting step. A second H2O2 molecule oxidizes the Mn2II to Mn2III, closing the proposed catalytic cycle.
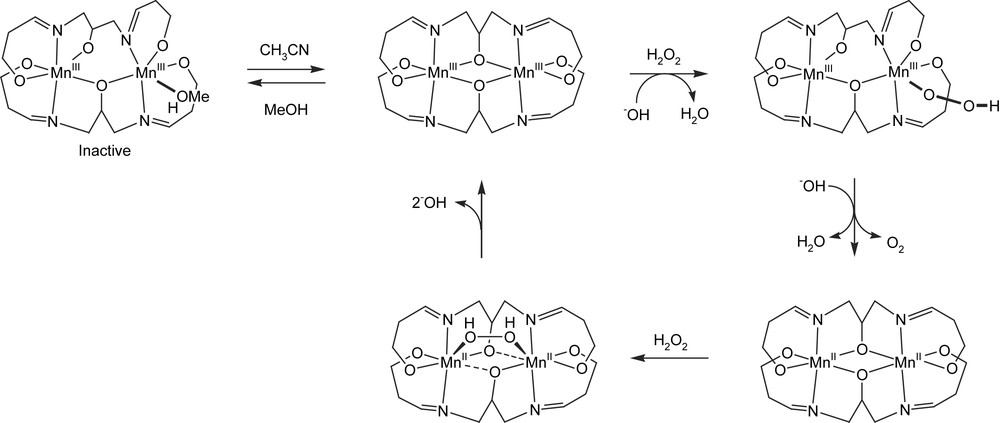
Proposed catalytic cycle for H2O2 decomposition by Mn–SALPN [33].
4 From catalase to catalysis – dinuclear carboxylate-bridged manganese catalysts
Whereas during catalase activity the dinuclear manganese complexes act as both an alternating 2e−reductant and oxidant of H2O2, for these manganese complexes to act as oxidation catalysts it is required that the oxidized state of the catalyst oxidizes the organic substrate instead of oxidizing H2O2. Indeed carboxylate-bridged dinuclear complexes based on a range of multidentate pyridyl, phenolate and amine based ligands, most notably those based on tmtacn [80–90], and tptn (N,N′,N″,N‴-tetrakis(2-pyridylmethyl)-1,3-propanediamine, Fig. 6) [91–94], have proven to be effective oxidation catalysts over the last several decades (Fig. 12) [95–100].
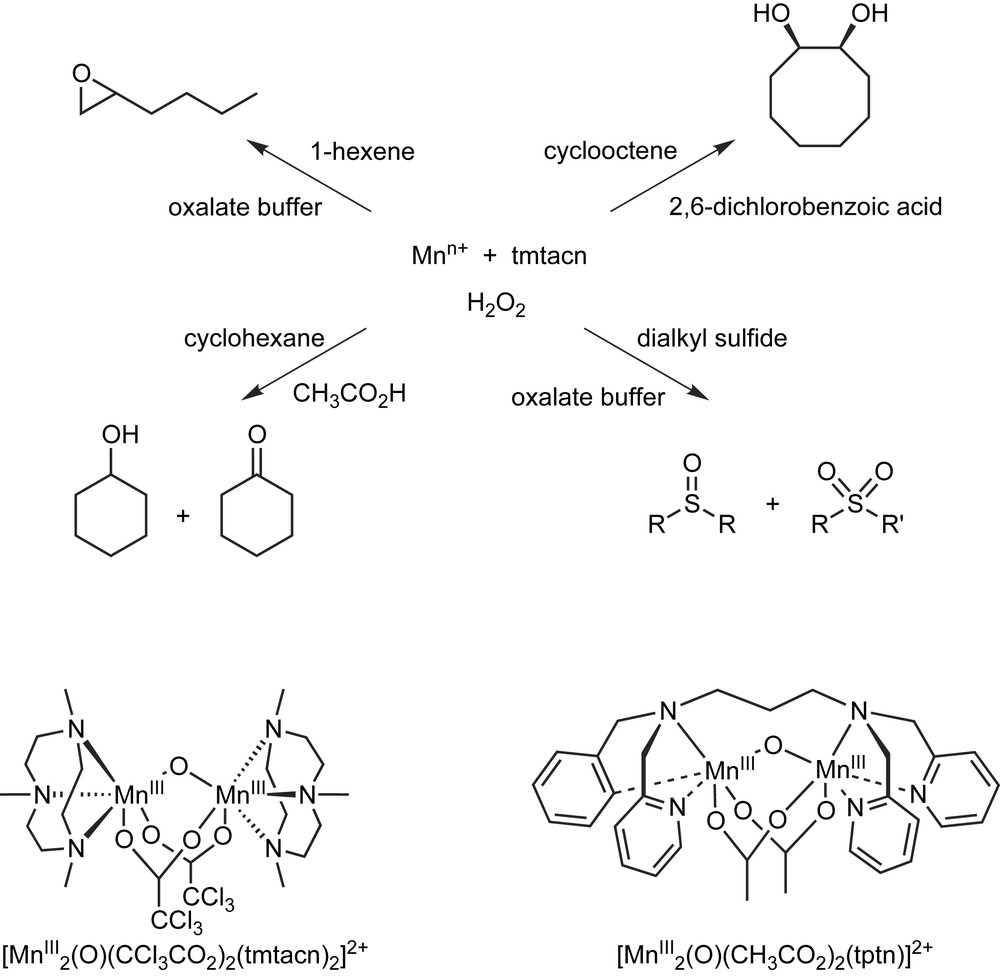
Examples of catalytic oxidation reactions.
This change in reactivity frequently involves only minor changes to the coordination environment of the dinuclear manganese core, exemplified in two different dinuclear complexes based on the same pyridyl/phenolate ligand (Fig. 13) [101,102]. Both complexes share similar coordination chemistry with a N2PyMePhOH ligand (2-{[[di(2-pyridyl)methyl](methyl)amino]methyl}phenol, Fig. 6) coordinating to each manganese centre and a bridging carboxylate ligand. However, if the phenolate ligands act as μ-O bridges between the manganese centres then the complex shows strong catalase activity whereas if the phenolates coordinate to only one of the metal centres and the manganese centres are bridged by a μ-hydroxy bridge then no catalase activity is observed, but instead the complex engages in the catalytic oxidation of organic substrates. The role of the carboxylate as a stable bridging ligand rather than a hemi-labile ligand has been demonstrated [102].
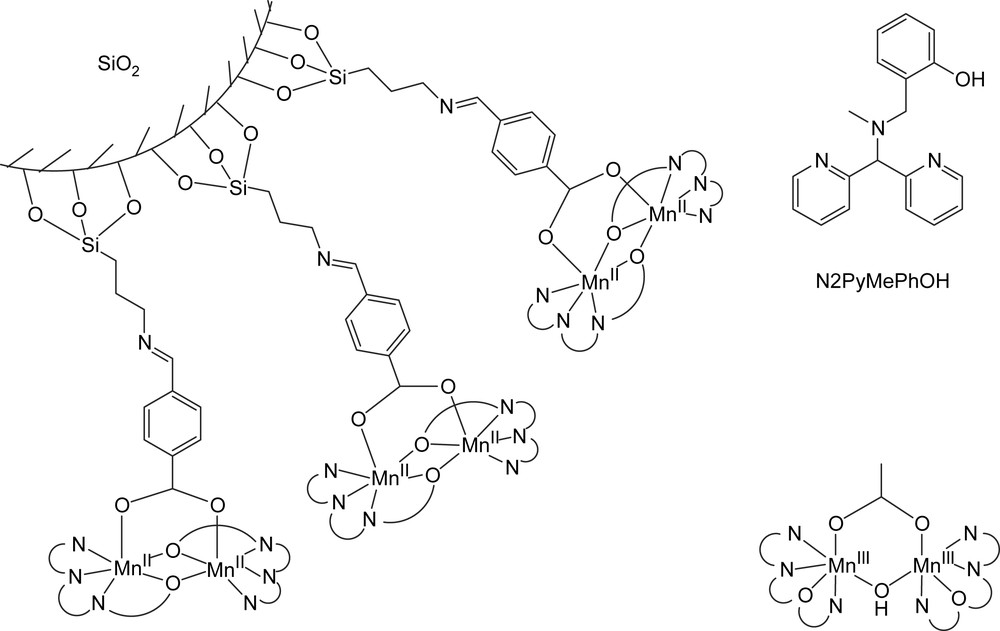
Surface bound catalase active Mn-dimers (left) and dinuclear [Mn2III(μ-OH)(N2PyMePhO)2]2+ active in epoxidation of styrene (right) [101,102].
Mn–tmtacn complexes have been employed extensively as oxidation catalyst for bleaching of laundry and for oxidative transformation of a wide range of organic substrates: epoxidation, cis-dihydroxylation, sulfoxidation, C–H activation and alcohol oxidation [96–98]. In many of these applications carboxylic acids are present in the reaction mixture (either as additive or as anion in the Mn-salt used). Although, to date a detailed understanding of the mechanism(s) by which these Mn–tmtacn catalysts operate is not available, it is apparent from recent work that the carboxylic acid additives are important as bridging ligands in the dinuclear [Mn2III(O)(RCO2)2(tmtacn)2]n+ complexes [80]. Indeed in this latter example the ligating role of the carboxylate ligand serves to control both reactivity and selectivity of the catalyst. Importantly with respect to the dinuclear manganese based catalase enzymes, for the tmtacn-based complexes the quantitative formation of bis-μ-carboxylate dinuclear complexes and the efficient consumption of H2O2 during catalysis suggest that the difference between catalase and oxidation catalysis activity may rest in very slight electronic differences.
5 Conclusions
Perhaps the most sweeping conclusion that may be drawn from the very many examples of carboxylate-bridged dinuclear manganese complexes that have been described to date is that the perception of the carboxylate bridging ligand simply as a labile/hemi-labile ligand, which can be conveniently displaced by hydrogen peroxide during both catalase activity and oxidation catalysis, is perhaps unwise. Indeed studies of enzymatic active sites provide strong support for the counter view that bridging carboxylate ligands serve to stabilize the dinuclear complexes during catalysis. In contrast to enzymatic and biomimetic catalase systems, the role of carboxylates in oxidation catalysis is at best unclear and in particular the nature of the active states in many manganese carboxylate based catalytic systems has proven somewhat elusive, in part, due to the reluctance to recognize the close relationship between catalase activity and hydrogen peroxide activation. It is apparent then that in order to understand the catalytic activity of manganese carboxylate oxidation systems, the role of the carboxylate as a non-labile bridging unit both in the resting and active states should be considered in addition to the more commonly postulated mononuclear high valent manganese-oxo species.
Acknowledgements
The authors thank Dr. Aurélie M. Brizard for assistance with translation. The Dutch Economy, Ecology, Technology (EET) programme (EETK01106), a joint programme of the Ministry of Economic Affairs, the Ministry of Education, Culture and Science, and the Ministry of Housing, Spatial Planning and the Environment is acknowledged for financial support.
1 Before the crystal structures of the (native, Cl− and N3−) inhibited enzymes were known, a similar catalytic mechanism was proposed, however, here initial μ-oxygen bridge opening is required to gain labile water ligand, e.g. Ref. [9].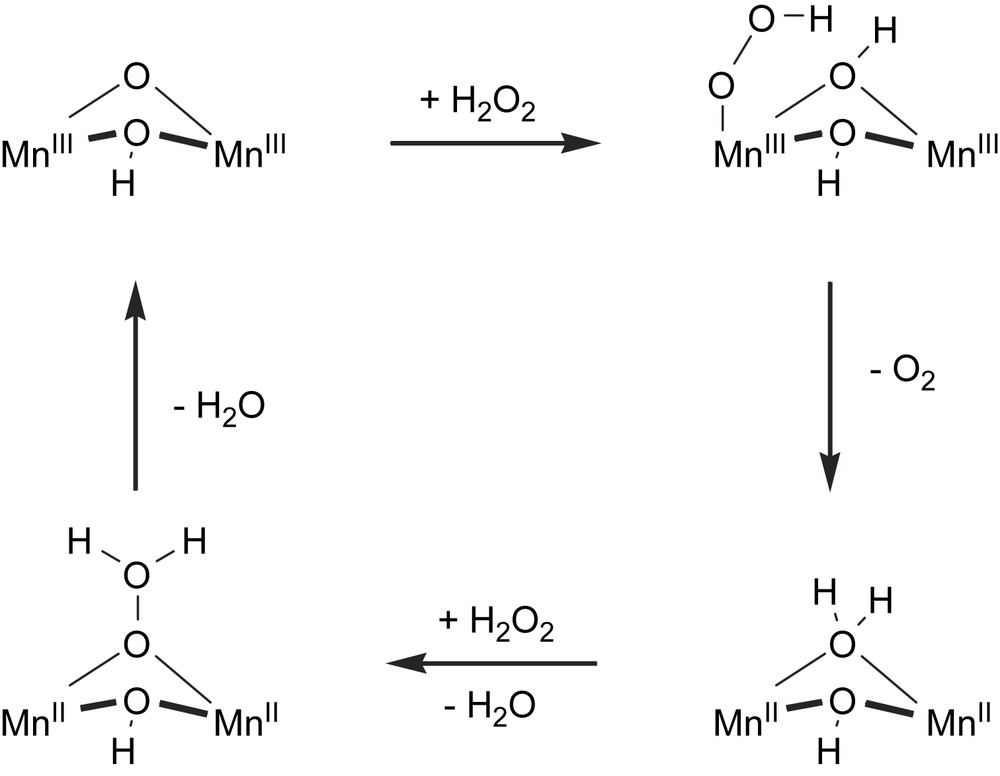
2 The phenol based systems of Sakiyama et al., despite containing carboxylate bridges, will not be discussed in detail, since their catalase activity is proposed to go via a Mn2III/Mn2IV cycle and thus do not resemble the natural enzymes (see Refs. [34–37]).
3 The latter is important, at least for the natural enzyme, since formation of the MnIIMnIII state in the presence of H2O2 eventually yields the kinetically inert superoxidized MnIIIMnIV. In other words, suppression of the formation of the mixed-valent MnIIMnIII state is essential for proper functioning of the enzymes. Hence, in addition to acting as a bridging ligand, the carboxylate bridge seems to play a key role in the enzyme by inhibiting one-electron processes and ensuring two-electron processes taking place under physiological conditions (see for example Ref. [9]).
4 See for example Refs. [85,86].
5 Although [Mn2III,IV(μ-O)2(bpia)2] is a structural model for the inactive Mn2III,IV state of the catalase enzyme, it does show catalase activity.
6 However, in the presence of 5 equiv. of hydroxide, an increase of the rate was observed with increasing water content of the reaction, see Ref. [30].
7 While a mixture of [Mn2IV(SALPN)2(μ-O)2] and [Mn2IV(3,5-Cl2-SALPN)2(μ-O)2] does not give rise to the formation of mixed ligands, however, when a mixture of both catalysts is used to decompose H2O2, mixed ligand species [Mn2IV(SALPN)(3,5-Cl2-SALPN)(μ-O)2] is formed. This, together with the observation that only 18O2 is formed for the decomposition of H218O2 using [Mn2IV(SALPN)2(μ-16O)2], indicates that the μ-oxo bridges are quite labile and monomers might be involved in the catalytic cycle (proposed to be via a Mn2IV/Mn2III cycle).