

1 Introduction
Oxidations of hydrocarbons with molecular oxygen and various donors of an oxygen atom (hydrogen peroxide, alkyl hydroperoxides, peroxy acids) are practically very important reactions since many industrial processes are based on these transformations [1]. As these processes use inexpensive non-poisonous oxidants and in many cases also environmentally benign solvents (for example, water), they belong to ‘green chemistry’ [2–7].
Such reactions can be considered as processes of C-H bond activation [8–10]. Generally, all these processes can be divided into three types. Reactions where organometallic derivatives (i.e., compounds containing an metal–carbon σ-bond) are formed as an intermediate or as the final product are assigned by us to the first type. The first group includes reactions involving ‘true’, ‘organometallic’ activation of the C–H bond. In the second group, we include reactions in which the contact between the complex and the C–H bond is only achieved via a complex ligand during the process of C–H bond cleavage, and the σ-C–M bond is not generated directly at any stage. In these reactions, the function of the metal complex usually consists in abstracting an electron or a hydrogen atom from the hydrocarbon. Finally, in the processes that belong to the third type, the complex does not initially activate the hydrocarbon, but the other reactant (for example, hydrogen peroxide or molecular oxygen). The reactive species formed (for example, hydroxyl radical) attacks then the hydrocarbon molecule without any participation in the latter process of the metal complex. The metal catalyst does not take part in the direct ‘activation’ of the C–H bond by the radical. From a mechanistic point of view, reactions described here belong to the second or/and third type.
2 Alkane oxidation: estimation of alkyl hydroperoxide content by GC analysis of the reaction solution samples before and after reduction with triphenylphosphine
In 1992, we described a very simple method to demonstrate [11–13] that H2O2 oxidations of alkanes catalysed by some transition metal complexes give substantial amounts of the corresponding alkyl hydroperoxides in addition to usually smaller concentrations of alcohols and ketones. This method (which we used in all our further works on alkane oxidations) is based on comparison of the chromatograms of the reaction solution made before and after the treatment of the sample with triphenylphosphine. If an excess of solid triphenylphosphine is added to a solution of alkane oxidation products 10–20 min before the GC analysis, the resulting chromatogram usually differs drastically from that of a sample not subjected to the reduction with PPh3. In the case of cyclohexane oxidation, the cyclohexanol peak rises markedly after the reduction while the intensity of the cyclohexanone peak decreases. Usually the sum of alcohol and ketone concentrations in the reduced sample is approximately equal to the total concentration of products in the solution untreated with triphenylphosphine. These results can be explained by the fact that the mixture of products of the reaction under investigation contains cyclohexyl hydroperoxide as the main component. Cyclohexyl hydroperoxide can totally decompose in the chromatograph to produce cyclohexanol and cyclohexanone. Alkyl hydroperoxides are known to be readily and quantitatively reduced by triphenylphosphine to yield corresponding alcohols. In the case of cyclohexyl hydroperoxide, the reduction gives cyclohexanol. After treatment of the reaction solution with PPh3, the GC analysis will give the amount of cyclohexanone that corresponds to the real concentration of this product in the reaction solution. The amount of cyclohexanol will give the sum of real concentrations of cyclohexyl hydroperoxide and cyclohexanol.
Thus, by comparing the data of chromatographic analysis of the reaction solution before and after reduction with triphenylphosphine, the amounts of cyclohexyl hydroperoxide really present in the solution at a given moment can be estimated quantitatively. One of the merits of this method is the possibility to estimate the concentration of the alkyl hydroperoxide formed from the alkane in the presence of an excess of an oxidant (hydrogen peroxide, alkyl hydroperoxide, peroxy acid or metal peroxide). It should be emphasized that the method described above is not a simple ‘quenching the reaction’ (indeed, works which determine alcohol and ketone concentrations only after addition of PPh3 give absolutely no information about the existence or non-existence of the corresponding alkyl hydroperoxides in the solutions).
3 Photo-induced metal-catalysed oxidations of alkanes by atmospheric oxygen
3.1 Photocatalysis by metal chlorides
In 90s we have described a new photocatalytic method for the transformation of alkanes and arylalkanes into corresponding hydroperoxides. Various metal chlorides were used as catalysts, but iron(III) chloride has been found to be the most efficient photocatalyst of alkane oxidation with atmospheric oxygen [14–22]. Halogenides of other transition metals CuCl2 [18, 21, 23, 24], AuCl4– [18, 21, 25, 26], PtCl62– [18, 26, 27], PtBr62–, RhCl3, RuCl3 [17] and CrCl3 + PhCH2NEt3Cl [28] also efficiently catalyse the aerobic oxygenation of alkanes in acetonitrile, methylene chloride or acetic acid under light irradiation. Methanol and formaldehyde were formed upon the slow bubbling of methane and air through a solution of HAuCl4 in MeCN under irradiation [25].
The proposed mechanism for the oxidation catalysed by iron(III) chloride includes as the first step the photo-excitation of the metal-containing species to stimulate homolysis of the Fe–Cl bond [22]. The chlorine radical attacks further the alkane. The formed iron(II) derivative can be oxidized either by molecular oxygen or the alkylperoxo radical.
Bromide complexes are also efficient in the photooxidation; however, in these cases the oxygenation of only alkylbenzenes (and not of alkanes) can be carried out. This can be due to lower reactivity of the bromine radical, which plays the role of an active species in this reaction. Mechanisms of the photooxygenation reaction in the cases of other than iron metal seem to be different from that postulated for the FeCl3-catalysed process [22]. Low-valent metal-containing species are apparently involved in the oxidation. It is interesting that the aerobic cyclohexane photooxidation in the presence of a catalytic system CrCl3–PhCH2NEt3Cl in MeCN produces not the corresponding cyclohexyl hydroperoxide but gives the ketone as a main product and only a small amount of the alcohol [28].
3.2 Photocatalysis by oxo complexes
In our earlier works, we demonstrated that the oxidation of alkanes and aromatic alkyl-substituted compounds by oxo derivatives of chromium(VI) is greatly accelerated by light irradiation [29–33]. Later we found that oxo compounds such as K2Cr2O7 in the two-phase solvent water-1,2-dichloroethane [34], CrO3 [35], complexes (n-Bu4N)2Cr2O7, (n-Bu4N)2Cr3O10 and (n-Bu4N)Cr4O13 [36], and UO2Cl2 [37] catalyse the photooxygenation of alkanes. Moreover, catalytic amounts of heteropolyoxometalates in methylene chloride, acetic acid, alcohols, acetone, acetonitrile [38, 39] transform alkanes and arylalkanes into the corresponding alkyl hydroperoxides as well as ketones and alcohols. Photooxygenation of alkanes catalysed by molybdenum or tungsten carbonyls begins apparently from the transformation of the carbonyl into an active oxo species [40, 41]. Some other chromium and vanadium complexes have been reported to be catalysts of alkane photooxidation [42–46]. The mechanism of aerobic alkane photooxidation catalysed by metal oxo complexes includes the formation of a photo-excited species, which is capable of abstracting a hydrogen atom from an alkane.
3.3 Photocatalysis by other complexes
The photooxidation of alkanes in acetonitrile occurs also in the presence of a catalytic amount of cyclopentadienyliron(II) complexes and bis(arene)iron(II) [47, 48]. The irradiation of an aqueous cyclohexane emulsion in the presence of an iron(III) salt, e.g. Fe(ClO4)3, gives rise [49] also to the formation of cyclohexanone instead of its mixture with cyclohexyl hydroperoxide and cyclohexanol. Palladium trifluoroacetate oxidizes alkanes under light irradiation [50].
3.4 Aerobic photooxidations catalysed by the ‘quinone-copper acetate’ system
Under the irradiation with visible light, an acetonitrile solution of an alkane and catalytic amounts of quinone and copper(II) acetate produces almost pure alkyl hydroperoxide, which decomposes only very slowly under these conditions [11, 51]. The first step of the reaction is a hydrogen atom abstraction from the alkane, RH, by a photo-excited quinone species to generate the alkyl radical, R•, and semiquinone. The former is rapidly transformed into ROO• and then into alkyl hydroperoxide, while the latter is re-oxidized by Cu(II) into the initial quinone.
4 Aerobic oxidations in the presence of a reducing agent
4.1 Catalysis by metaloporphyrins
Porphyrin complexes of some transition metals are used as models of heme monooxygenases. The porphyrin ring can play the role of an electron donor owing to its ability to generate the radical cation and thus become an active participant of the catalytic process. If molecular oxygen is the oxidant in model reactions, a reducing agent is required whose role can be played, for example, by metallic zinc, thiosalicylic acid or molecular hydrogen. We employed in our investigations ferrocene and benzylferrocene as the reductant and tetraphenylporphyrinate complexes of cobalt(II) or iron(III) [52]. Alkane oxidation in the presence of air and an electron source occurs at normal temperature and pressure. In the cyclohexane oxidation, cobalt-containing catalyst turned out to be more efficient than iron porphyrin, and the product yield was higher when benzylferrocene was used instead of ferrocene.
4.2 Catalysis by complexes of vanadium, gold and manganese
Metal porphyrinates can be replaced in biomimetic studies by simple salts of transition metals. We have found that FeCl3, CuCl2 and NaAuCl4 induce aerobic oxygenation of benzene, ethylbenzene, styrene, cyclohexane and n-hexane in aqueous acetonitrile if ascorbic acid is added to the solution [53]. However, unfortunately turnover numbers (moles of products per one mole of the catalyst) did not exceed 3. When d-glucose was used instead of ascorbic acid, only ethylbenzene have been oxygenated, CuCl2 exhibited the highest activity.
Vanadium is known to play the important role in biology. Some complexes have been used as models of natural vanadium-containing compounds (see, for example, [54]). Recently we demonstrated [55] that vanadium complexes (particularly vanadate anion) catalyse aerobic hydroxylation of benzene to phenol in acetonitrile in the presence of solid ascorbic acid and with obligatory participation of pyridine, pyrazine-2-carboxylic acid and acetic acid as mediators of proton and electron transfer. If some water is added to the system containing ascorbic acid, this compound is dissolved in aqueous acetonitrile and no hydroxylation occurs. The oxidation of toluene under the same conditions gave a mixture of isomeric cresols with the ratio o:m:p = 60:16:24. Naphthalene gave isomeric naphthols (α:β=4:1). Ascorbic acid can be replaced with zinc powder. The zinc-based system hydroxylates not only benzene and aromatic ring and methyl group in toluene (o:m:p = 47:28:25) but also C–H bonds in cyclohexane with total turnover number being 78. A mechanism of the formation of hydroxyl radicals by these systems has been proposed (Fig. 1). Initial vanadium(V) species, conventionally depicted in the scheme as 1, is reduced to a catalytically active V(IV) form 2 by ascorbic acid or Zn0. The latter species adds molecular oxygen and after reduction is converted to a hydroperoxy V(IV) species (5). A homolytic splitting O–O bond in this species gives rise to more stable oxo derivative of V(V), 6, and hydroxyl radical. Finally, step 6 leads to the initial catalytically active species 1. In this sequence of stages, PCA can facilitate proton transfer, for example intermolecular transfer between two HO groups or between proton donors from the reaction media and oxo or hydroxy groups at the vanadium ion. Protonated pyridine apparently plays the role of a hydrogen atom mediator and is a model of NADH. Reduction of V(V) by NADH in biological systems is known [56, 57]. Thus we can conclude that these systems mimic generation of hydroxyl radicals in certain vanadium-dependent biological processes.
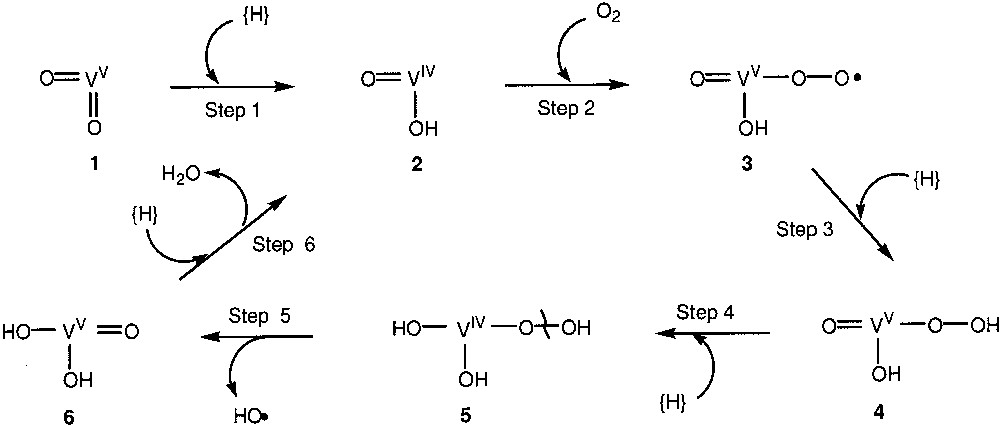
Proposed mechanism of the formation of hydroxyl radicals.
The oxidation of cyclooctane by air at room temperature catalysed by NaAuCl4 in the presence of Zn/CH3COOH as a reducing agent and methylviologen as electron-transfer agent gave cyclooctanol (TON = 10) [58]. A binuclear Mn(IV) complex with the 1,4,7-trimethyl-1,4,7-triazacyclononane macrocyclic ligand (compound 21; see below) initiates oxidation of cyclohexanecarboxaldehyde with atmospheric oxygen in acetonitrile under heating resulting in the formation of cyclohexanecarboxylic acid and considerable amounts of oxidative decarboxylation products, i.e., cyclohexane, cyclohexanol and cyclohexanone [59]. In the presence of cyclooctane, the reaction gives rise also to the formation of cyclooctanol and cyclooctanone.
4.3 Dioxygenase-like vanadium-catalysed splitting C–C bonds
Brégeault and co-workers have shown that vanadium complexes catalyse aerobic oxidation of ketones with splitting C-C bonds; particularly, 2-hydroxycyclohexanone can be easily oxidized to adipic acid [60, 61]. Very recently, we used the mononuclear anionic dioxo vanadium (V) species, [{Ph3SiO}2VO2]–, and the dinuclear complex anion, [{Ph2SiO2VO2}2]2–, as well as simple vanadium derivative, n-Bu4NVO3, VOSO4·5 H2O, VO(acac)2, H6PMo9V3O40 as catalysts for the aerobic C–C-bond cleavage in 2-hydroxycyclohexanone and of 2-methylcyclohexanone [62]. On the basis of the obtained data, a mechanism has been proposed, which is depicted in Fig. 2 for the case of C–C-bond cleavage in 2-hydroxycyclohexane. This scheme presents a formally closed catalytic cycle, although we realise that in reality some other steps take place and the equilibria are more complex. The reaction can begin from the formation of catalytically active oxovanadium(V) species 6, which is depicted as [VO2]+X–. Certainly this species contains some additional ligands such as coordinated water from the organic solvent. Species 6 can be coordinated to the substrate molecule via its hydroxyl oxygen to produce complex 7. Proton transfer from the carbon atom to the oxo group leads to the formation of the enol form (8). Naturally, this proton transfer does not occur in an intramolecular way, as it is schematically shown as Step 2, but via deprotonation and protonation, with participation of the solvent or water (which was present in relatively small concentrations in all reaction solutions). Step 3 is a single electron transfer from the electron-rich organic moiety to the V(V)-oxo part of the complex to produce species 9, which contains the n-coordinated to V(IV) organic part of the complex. There is an unpaired electron on the organic ligand, and one of the mesomeric forms of 9 can be a carbon-centred radical. In the presence of molecular oxygen species 9 will react rapidly with O2. Indeed, it is well known [8] that carbon-centred radicals add to the oxygen molecule in a very fast reaction:
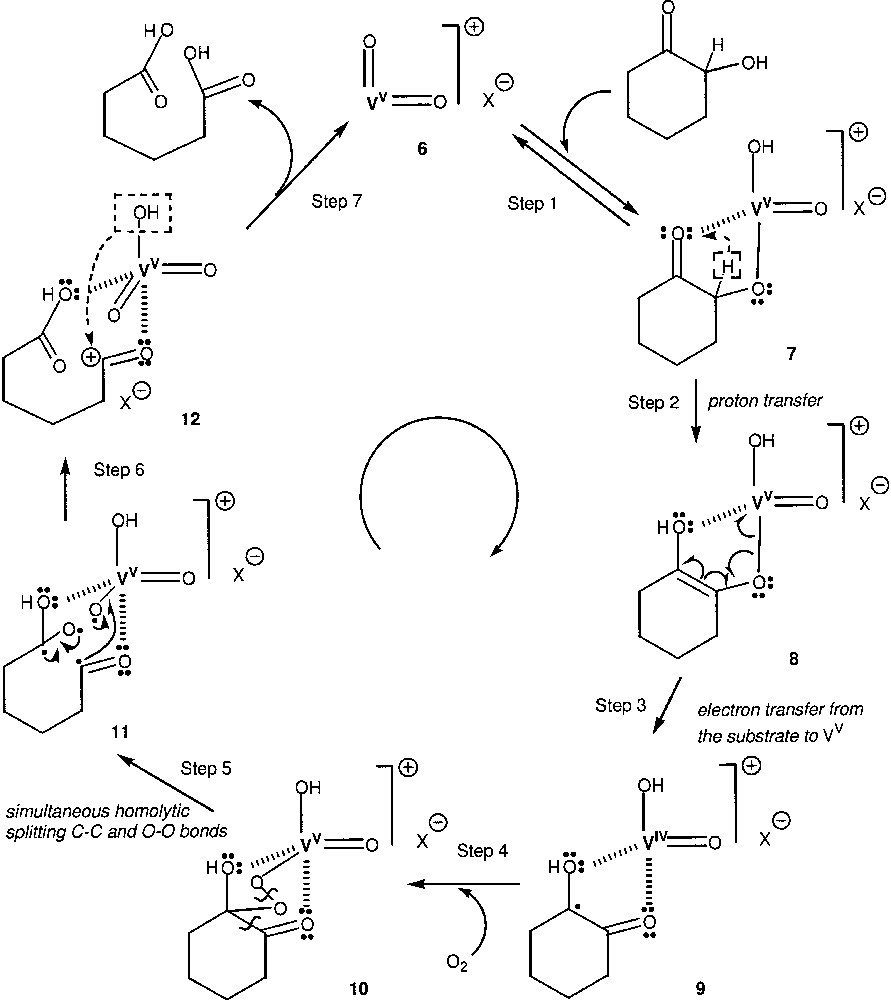
Proposed mechanism for the C–C-bond cleavage in 2-hydroxycyclohexane.
A similar mechanism has been proposed previously for the oxidative C–C bond splitting in coordinated to palladium(II) acetylacetonate ligand to afford acetate ligand [8, 63]. In that case, the reaction proceeded smoothly at room temperature under irradiation by visible light. Another ligand, the coordinated to Pd(II) ortho-metalated azobenzene played a role of an ‘antenna’, absorbing the light energy necessary for the electron transfer from the acac ligand to the palladium centre. It should be noted that such a type mechanism is relative to the mechanism of C–C bond splitting in substituted phenols and catechols under the action of O2, catalysed by enzymes non-heme iron dioxygenases (see, for example, [64–67]). Thus oxidative systems based on vanadium complexes can be considered as biomimetic models of dioxygenases.
5 Systems based on hydrogen peroxide
Hydrogen peroxide is a relatively inexpensive reagent; it is not toxic. This compound readily transfers one of its oxygen atoms to a substrate to produce as a by-product a ‘green’ substance, water, which is a great advantage of hydrogen peroxide in comparison with all other oxidants (the sole exception is molecular oxygen or air). We have discovered a few catalytic systems based on hydrogen peroxide as oxidant, some of which are very efficient. Mechanistic studies (kinetics, selectivities etc.) showed that these systems oxidize alkanes and other organic compounds via different mechanisms. Thus the ‘O2–H2O2–vanadium complex-pyrazine-2-carboxylic acid’ reagent unambiguously generates hydroxyl radicals, whereas the ‘[L2Mn2O3](PF6)2 (L = 1,4,7-trimethyl-1,4-7-triazacyclononane)-carboxylic acid’ system oxidizes apparently with the formation of the MnV=O species. Mechanisms of some other reactions are unclear.
5.1 The ‘O2–H2O2–vanadium complex–pyrazine-2-carboxylic acid’ reagent
Any soluble vanadium derivative [vanadate anion in the form n-Bu4NVO3, or VOSO4, VCl3, VO(acac)2] can be used as a catalyst in combination with pyrazine-2-carboxylic acid as co-catalyst for the oxidations with hydrogen peroxide in acetonitrile solution [12, 68–83]. At low temperatures, the predominant product of the alkane oxidation is the corresponding alkyl hydroperoxide (alcohols and ketones or aldehydes are formed simultaneously in smaller amounts). This alkyl hydroperoxide then slowly decomposes to produce the corresponding ketone and alcohol. The amounts of alkyl hydroperoxide, alkanol and alkanone were estimated by comparing the data of chromatographic analysis of the solution before and after reduction with triphenylphosphine (see above), as well as by direct measure of the intensities of the peaks corresponding to ROOH. Atmospheric oxygen takes part in this reaction; in the absence of air, the oxygenation reaction does not proceed. Thus it may be concluded that in alkane oxidation, hydrogen peroxide plays the role of a promoter, while atmospheric oxygen is the true oxidant. The oxidation of n-heptane by the reagent under consideration exhibits low selectivity, C(1):C(2):C(3):C(4) ≈ 1:4:4:4. This parameter is close to that found for the oxidation of n-heptane by H2O2 in MeCN under UV irradiation (1.0:3.4:3.2:3.0). The selectivities of the reactions with branched alkanes (2- and 3-methylhexane, cis- and trans-decalin) are also very similar to those observed with hydroxylation of the alkanes with hydrogen peroxide under UV irradiation. Methane, ethane, propane, n-butane and isobutane can be also readily oxidized in acetonitrile by the same reagent. Alcohols, aldehydes or ketones and carboxylic acids are obtained with high total turnover numbers (at 75 °C after 4 h: 420 for methane and 2130 for ethane) and H2O2 efficiency in addition to the primary oxidation products (alkyl hydroperoxides). Methane can also be oxidized in aqueous solution, giving in this case methanol as the product (after 20 h at 20 °C, the turnover number equals 250). The reagent also oxygenates arenes to phenols, alcohols to ketones and hydroperoxidizes the allylic position in olefins.
The crucial step of the oxidation by the ‘O2–H2O2–VO3––pyrazine-2-carboxylic acid’ reagent is the very efficient generation of HO• radicals. These radicals abstract hydrogen atoms from the alkane, RH, to generate the alkyl radicals, R•. The latter react rapidly with an O2 molecule affording the peroxo radicals, ROO•, which is then transformed simultaneously into three products: alkyl hydroperoxide, ketone, and alcohol. The proposed mechanism (Fig. 3) of HO• generation involves the reduction of V(V) species 13 by the first molecule of H2O2 to give V(IV) derivative 16. No oxidation occurs in the absence of pyrazine-2-carboxylic acid. The possible role of pyrazine-2-carboxylic acid is its participation (in the form of a ligand at vanadium centre) in the proton transfer, which gives the hydroperoxy derivative of vanadium. One can assume that the nitrogen atom of this ligand is de-coordinated and then abstracts a proton from the coordinated hydrogen peroxide molecule. As a result, a pyridinium base is formed, which, after rotation of the ‘robot’s arm’, approaches one of the =O ligands at vanadium and protonates it to give the –OH ligand (Fig. 4).
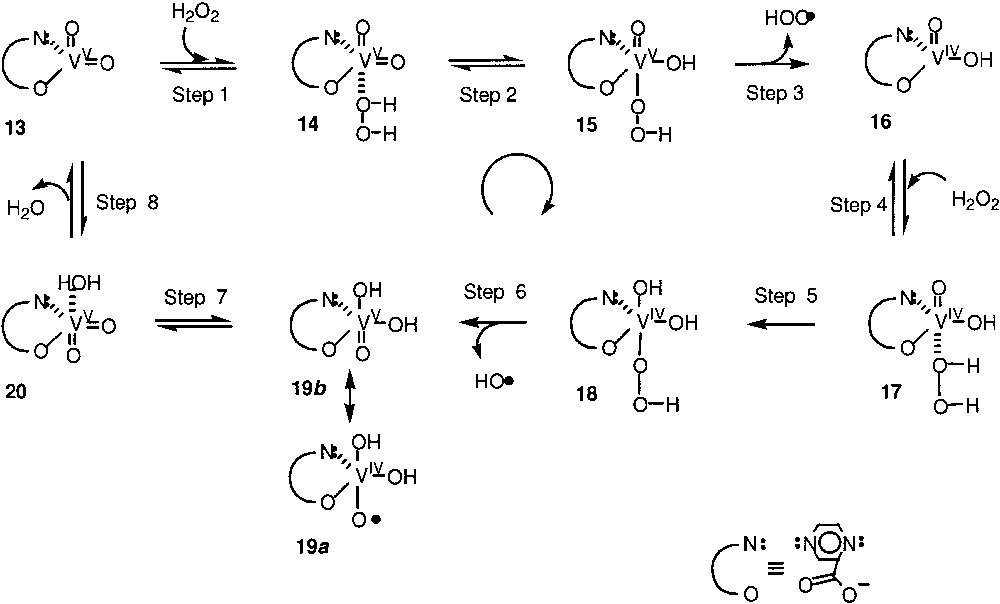
Proposed mechanism of HO• generation involving the reduction of V(V) species 13 by the first molecule of H2O2 to give V(IV) derivative 16.
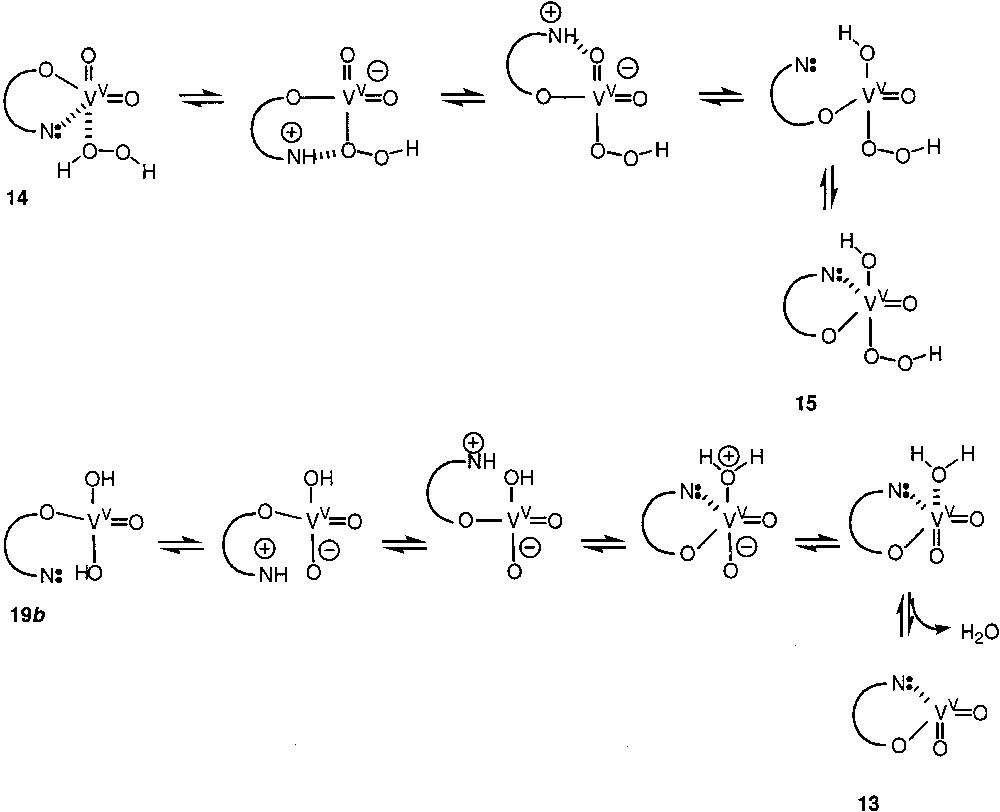
Formation of a pyridinium base is formed, which, after rotation of the ‘robot’s arm’, approaches one of the = O ligands at vanadium and protonates it to give the –OH ligand.
Alkanes can be oxidized by hydrogen peroxide in acetonitrile using tetra-n-butylammonium salts of the vanadium-containing polyphosphomolybdate [PMo11VO40]4– as catalyst [84]. The oxidation of alkanes gives rise to the corresponding alkyl hydroperoxides as the main products, which slowly decompose in the course of the reaction to produce the corresponding ketones (aldehydes) and alcohols. The total yield of the reaction products in the presence of pyrazine-2-carboxylic acid is almost unchanged; however, the initial reaction rate is higher in this case.
5.2 The ‘[L2Mn2O3](PF6)2 (L = 1,4,7-trimethyl-1,4,7-triazacyclononane)-carboxylic acid’ system
We have demonstrated that manganese (IV) derivative [L2Mn2O3](PF6)2 (21) (L = 1,4,7-trimethyl-1,4-7-triazacyclononane) (Fig. 5) catalyses very efficiently the oxygenation of various organic compounds in acetonitrile or nitromethane, only if a carboxylic acid is present in small concentrations in the reaction mixture [85–90]. Higher (n-hexane and n-heptane, decalin, cyclohexane, methylcyclohexane etc.) and light (methane, ethane, propane, normal butane and isobutane) alkanes can be easily oxidized by this system at room temperature, at 0 °C and even at –22 °C. Turnover numbers of 3300 have been attained and the yield of oxygenated products is 46%, based on the alkane. The oxidation initially affords the corresponding alkyl hydroperoxide as the predominant product; however, this compound decomposes later on, to produce the corresponding ketone and alcohol.
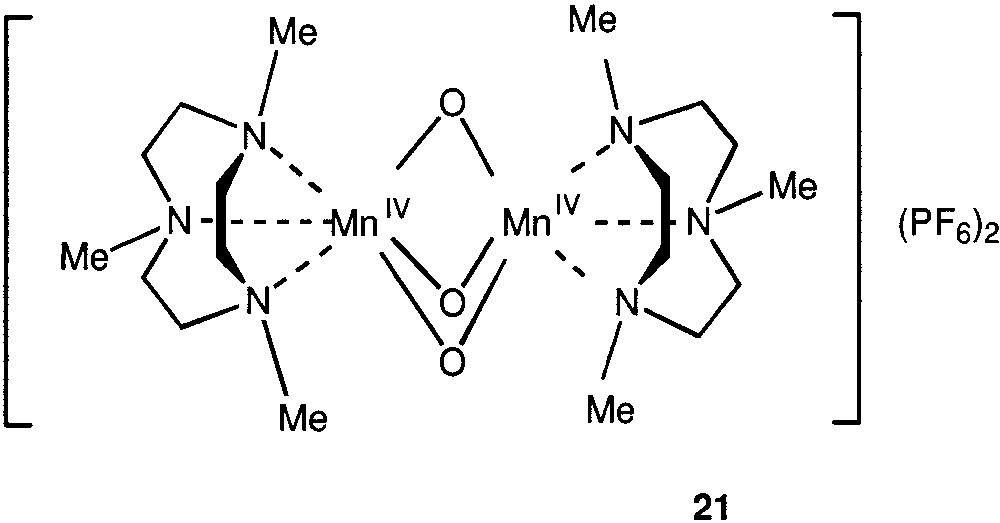
Compound 21.
Regio- and bond-selectivities of the reaction are high: C(1):C(2):C(3):C(4) ≈ 1:40:35:35 and 1°:2°:3° is 1:(15–40):(180–300). The reaction with cis- or trans-isomers of decalin gives (after treatment with PPh3) alcohols hydroxylated in the tertiary position with a cis/trans ratio of ~2 in the case of cis-decalin, and with a trans/cis ratio of ~30 in the case of trans-decalin. It has been proposed [89] that the catalytically active compound 24 is formed in the solution (Fig. 6). The alkane oxidation in the oxidase cycle begins with hydrogen-atom abstraction from the alkane by an oxygen-centred radical or a radical-like species 28 (Fig. 7). The active oxidant is probably a dinuclear manganese complex (HOO–)MnMn(=O), and the reaction occurs via an ‘oxygen-rebound mechanism’ between radical R• and HOO- group, to produce ROOH with retention of stereochemistry (step 10). Alkyl radicals (R•) can also partially escape from the solvent cage and react with dioxygen to generate ROO• and subsequently ROOH, with some loss of stereochemistry. Species 24 also simultaneously catalyses the ‘non-productive’ H2O2 decomposition to O2 and H2O in the catalase cycle (Fig. 7).
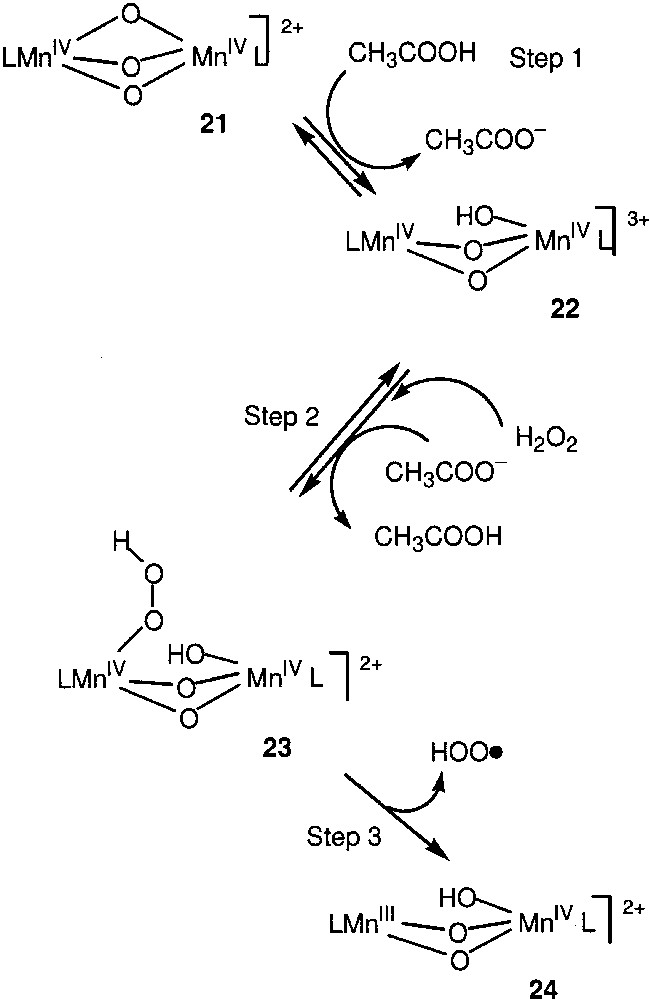
Formation of the catalytically active compound 24.
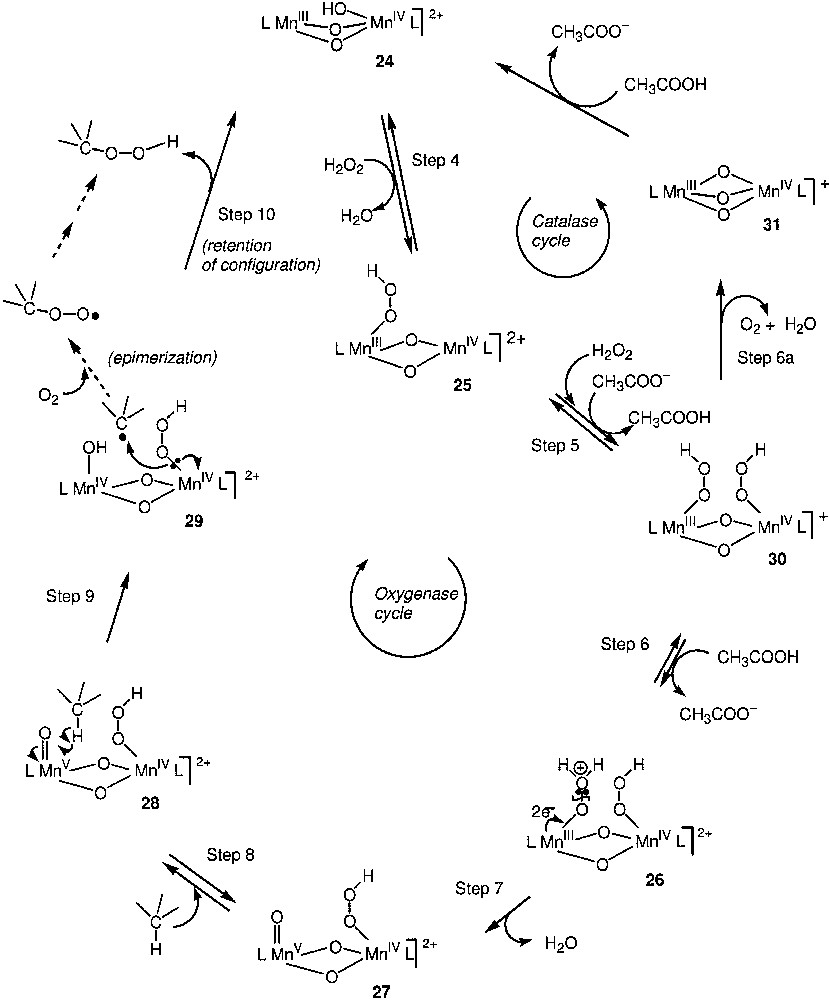
Alkane oxidation in the oxidase cycle, beginning with hydrogen-atom abstraction from the alkane by an oxygen-centred radical or a radical-like species 28.
Soluble manganese (IV) complex containing as ligands 1,4,7-triazacyclononane moieties bound to a polymeric chain (compound 32, Fig. 8) catalyses [90] the oxidation of alkanes by hydrogen peroxide in acetonitrile at room and lower temperatures. Corresponding alkyl hydroperoxides are the main products. The presence of relatively small amount of acetic acid is obligatory for this reaction. The oxidation of alkanes and olefins exhibits some features (kinetic isotope effect, bond selectivities) that distinguish this system from an analogous system based on dinuclear Mn(IV) complex 21.
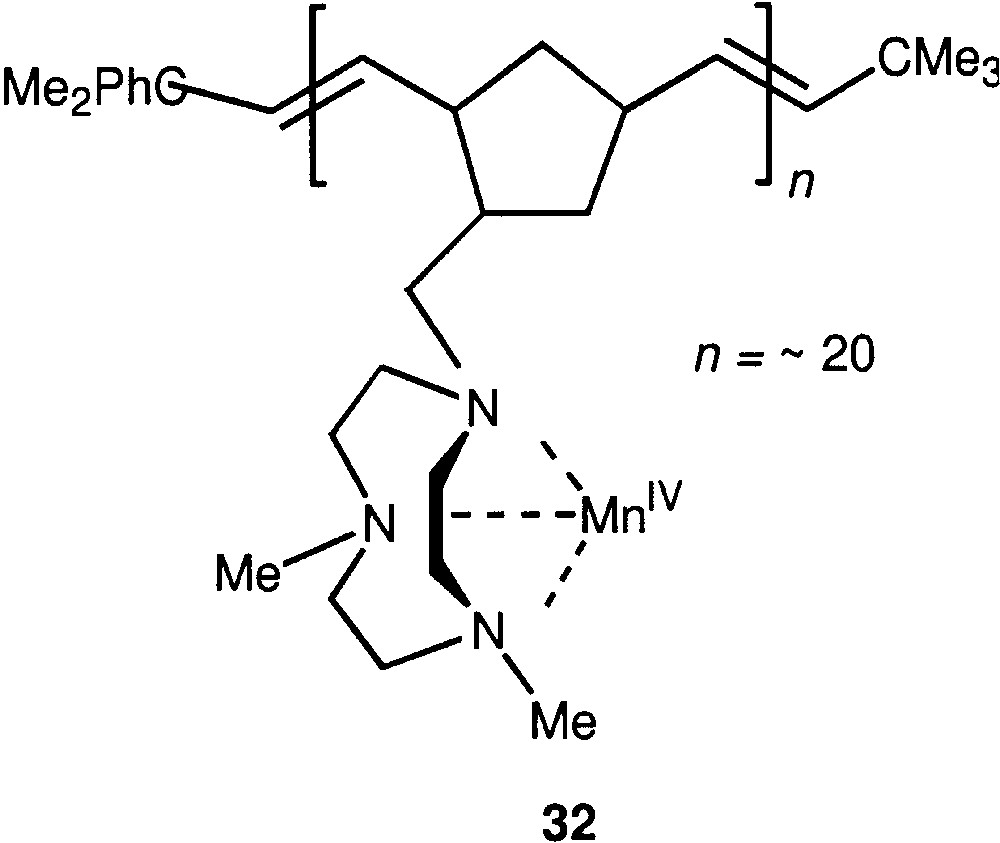
Compound 32.
The ‘H2O2–compound 21–MeCO2H’ system also [88] transforms secondary alcohols into the corresponding ketones with quantitative yields at room temperature within a few minutes; the yields of aldehydes and carboxylic acids in the oxidation of primary alcohols are lower. Terminal aliphatic olefins such as hexene-1 are quantitatively epoxidized by the same system in acetonitrile at room temperature within 20 min, while the epoxide yield in the analogous reaction with styrene attains only 60% under the same conditions. Finally, dimethylsulfide can be quantitatively and selectively converted into dimethylsulfoxide within 3 h at room temperature.
5.3 Hydrogen peroxide oxidations catalysed by other metal complexes
5.3.1 Osmium compounds as catalysts
Osmium derivatives, especially OsCl3, catalyse the oxygenation of saturated hydrocarbons with hydrogen peroxide [91, 92]. For example, the reaction with cycloheptane at 80 °C gave, after 3 h, cycloheptanol and cycloheptanone, the total turnover number (TON) being 63. Addition of small amount of pyridine gave rise to a noticeable increase of the yield and to the predominant formation of the ketone (TON = 112 after 90 min). The reaction is accompanied by non-productive H2O2 decomposition to give molecular oxygen. By varying the additive, it is possible to control the ketone/alcohol ratio. Lower alkanes can be also easily oxidized into MeCN, if pyridine is added to the reaction solution. In contrast to methane, which affords methanol as the main product, ethane and propane give mainly carbonyl compounds and only smaller amounts of alcohols, with TON values of 102 and 150, respectively.
The selectivities of the alkane oxidations catalysed by OsCl3 are higher than those determined for analogous hydroxylations by the ‘H2O2–hν’ and ‘H2O2–FeSO4’ systems as well as ‘H2O2–VO3––pyrazinecarboxylic acid’ reagent (see above), which are believed to produce free hydroxyl radicals. It is noteworthy that selectivities significantly increase when pyridine is added to the reaction solution. Moreover, while the oxidation of cis-decalin with H2O2–OsCl3 in MeCN occurs without retention of the configuration (the trans/cis ratio of the formed products being even more than unity), in the presence of pyridine the reaction becomes more selective, the trans/cis ratio decreasing with increasing pyridine concentration. It is important to note that in MeCO2H the reaction exhibits stereoselectivity (the value of the trans/cis parameter was 0.26). This value of the trans/cis parameter for the ‘H2O2–OsCl3’ system in MeCO2H is only slightly higher than that for the hydroxylation in MeCN by the ‘H2O2–21–MeCO2H’ system (see above).
It has been proposed that the oxidation by the H2O2–OsCl3 system starts with the hydrogen atom abstraction from the alkane by an oxo-osmium complex, and the reaction occurs in a solvent cage. The alkyl radicals formed react then with dioxygen to generate the corresponding alkyl hydroperoxide, which decomposes to afford the corresponding ketone and alcohol. Coordination of a nitrogen-containing heterocycle to osmium may produce a catalytically active oxo species surrounded with voluminous ligands, and the relatively high bond- and stereo-selectivities of the alkane oxidation may be due in this case to the bulkiness of the ligands at the reaction centre.
5.3.2 Catalysis by chromium(VI) compounds
It has been shown in our earlier work [93, 94] that cyclohexane can be oxidized by hydrogen peroxide at room temperature when a Cr(VI) oxocomplex is used as the catalyst; however, the product yields were very low. More recently, we found [95, 96] that chromic acid catalyses the oxidation of the alkanes with hydrogen peroxide under mild conditions; the reaction is particularly efficient for the very inert ethane: CH3CH3 is oxidized by H2O2 in acetonitrile solution containing catalytic quantities of H2CrO4 at 60 °C to give ethyl hydroperoxide, acetaldehyde, ethanol and acetic acid, the total turnover number being 620 after 1 h; the maximum yield equals 21% based on H2O2. The relative content of these products depends on the initial concentration of hydrogen peroxide; at its high concentrations, acetaldehyde becomes a predominant product.
The main product of cyclooctane oxidation is the alkyl hydroperoxide, which gradually decomposes to give predominantly cyclooctanone. The reaction is accelerated but insignificantly (ca 15%) if pyridine is added. After 8 h, the turnover number had attained 280. The oxidation with H2O2 in CH3COOH as a solvent is less efficient. Selectivity parameters for the oxidation of decalin (cis- and trans-isomers) and adamantane are different from those obtained for HO-radical-generating systems. The Cr-containing systems oxidize non-stereoselectively. It has been concluded that the Cr-catalysed reactions does not seem to proceed via HO radicals, but involve possibly peroxo complexes as oxidizing species.
5.3.3 Oxidations catalysed by derivatives of nickel, platinum, rhenium and gold
If nickel(II) perchlorate is used as a catalyst and hydrogen peroxide as an oxidant, the reaction of cyclooctane and other alkanes in acetonitrile solution at 70 °C gave alkyl hydroperoxides as main products, whereas concentrations of the corresponding alcohols and ketones were much lower [97]. The hydroperoxidation of cyclooctane proceeds very slowly, with a significant auto-acceleration after 50 h. It is interesting to note that addition of a small amount of 1,4,7-trimethyl-1,4,7-triazacyclononane dramatically enhances the reaction rate at the beginning of the oxidation, whereas the addition of picolinic acid instead of TMTACN almost completely depresses the cyclooctane oxidation. The oxidations by the H2O2–Ni(ClO4)2–TMTACN system exhibit very low selectivities for n-heptane and branched alkanes and the reaction with cis-decalin is not stereoselective. These data testify clearly that Ni(II)-catalysed alkane oxygenation proceeds mainly with participation of free hydroxyl radicals.
We have also investigated the alkane oxidation with H2O2, catalysed by H2PtCl6 [97]. In comparison with the analogous Ni(II)-catalysed process, this reaction proceeds much more rapidly and concentrations of relatively stable cyclooctanol and cyclooctanone are high. The turnover number attains 44 after 44 h. The reaction catalysed by Pt(IV) exhibits relatively high selectivities for branched alkanes. It has been concluded on the basis of the selectivity parameters that the alkane oxidation catalysed by Pt(IV) at least in one of the possible routes involves not free hydroxyl radicals but oxo or peroxo complexes of platinum.
Pyrazine-2-carboxylic acid accelerates the hydrocarbon oxidation catalysed by CH3ReO3 [98]. Gold(III) and gold(I) complexes, NaAuCl4 and ClAuPPh3 also catalyse efficiently the oxidation of alkanes by H2O2 in acetonitrile solution at 75 °C [58]. Turnover numbers attain 520 after 144 h. Alkyl hydroperoxides are the main products, whereas ketones (aldehydes) and alcohols are formed in smaller concentrations. At least one of the pathways in Au-catalysed alkane hydroperoxidation does not possibly involve the participation of free hydroxyl radicals and the oxidation begins from the alkane hydrogen atom abstraction by a gold oxo species.
5.3.4 Biomimetic oxidations with participation of dinuclear iron complexes
Various dinuclear iron complexes have been synthesized as structural models of diiron non-heme enzymes and their properties were studied. In some cases, activity of such complexes in saturated and aromatic hydrocarbon oxygenations was reported. We have found [99] that, when used in relatively low concentration, compound 33 (Fig. 9) does not catalyse cyclohexane oxidation with H2O2 in acetonitrile at room temperature. However, when certain amino acids (pyrazine-2-carboxylic or pyrazine-2,3-dicarboxylic acid) are added in low concentration, efficient cyclohexane oxygenation can be noticed. For the case of pyrazine-2-carboxylic acid, TON = 140. The efficiency of pyrazine-2,3-dicarboxylic acid as co-catalyst is somewhat lower under exactly the same conditions.
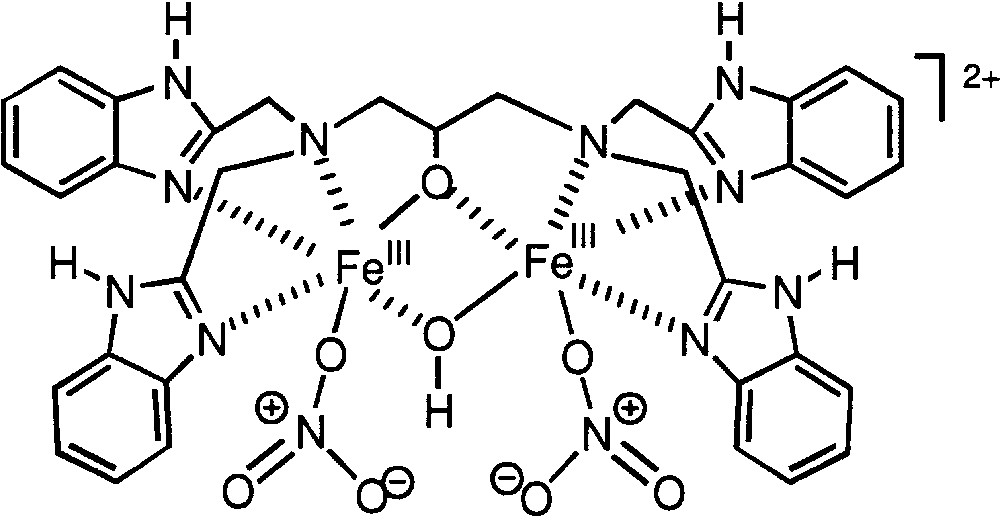
Cation of compound 33.
The cyclohexane oxygenation reaction is of a first order with respect to both hydrogen peroxide and alkane concentration, since the initial rate of the reaction is proportional to [H2O2]0 and [cyclohexane]0, respectively. Ethane was transformed by this system mainly to ethyl hydroperoxide as well as to acetaldehyde, in a minor amount. Acetic acid in a very small concentration was detected after 6 h. After this time, the concentrations of ethyl hydroperoxide and acetaldehyde corresponded to TON = 21. Methane was oxidized at 25 °C to produce, after 6 h, methyl hydroperoxide and formaldehyde with TON = 4.
The first step of the process is the reduction of one FeIII ions with a hydrogen peroxide molecule to produce hydroperoxy radicals and FeII. The interaction of FeII with H2O2 possibly begins from the formation of hydroperoxy derivative. It was assumed that amino acids added to the reaction mixture play a very important role in this stage, facilitating the proton transfer between the coordinated H2O2 molecule and ligands at iron centres. Hydroxyl radical attacks an alkane molecule and the alkyl radical thus formed adds rapidly an oxygen molecule affording the corresponding alkyl peroxy radical. This radical can be reduced by one of the two iron(II) centres in a dinuclear complex and, after addition of a proton, a molecule of the alkyl hydroperoxide is formed.
Very recently, we found [100] that hydrogen peroxide oxidizes alkanes in acetonitrile at room temperature if dinuclear iron complex with 1,4,7-triazacyclononane (TACN), compound 34 (which is a model for O2-transporting proteins hemerythrins), is used as a catalyst (Fig. 10). Pyrazine-2-carboxylic acid accelerates the oxidation [100].
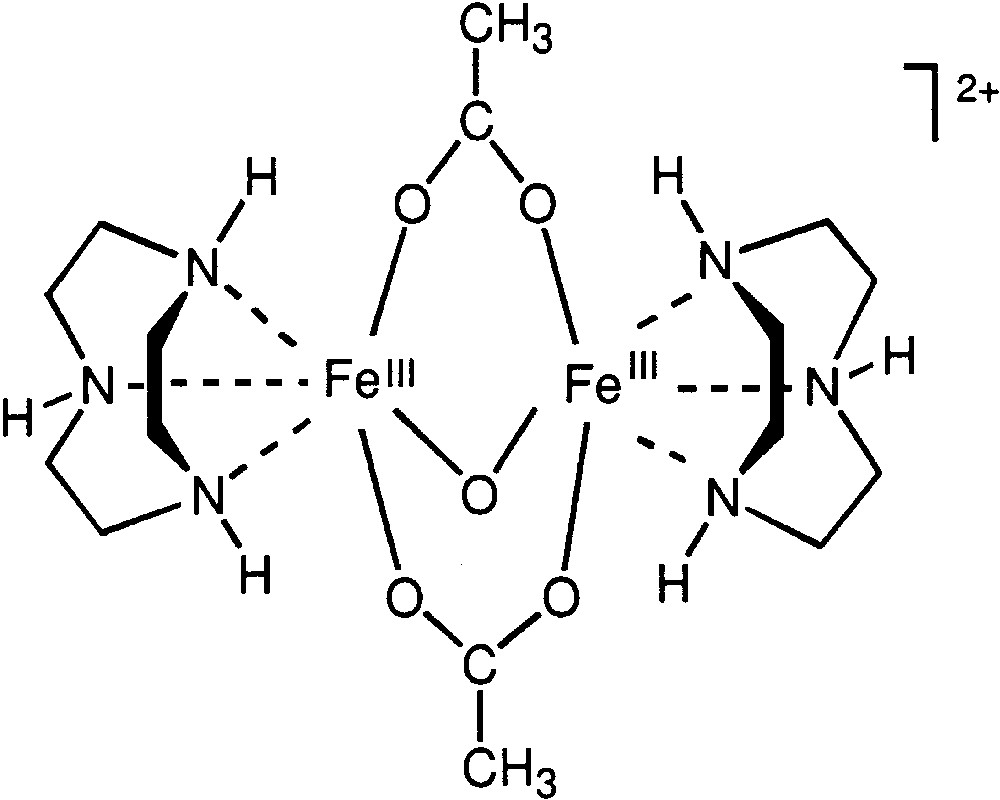
Cation of compound 34.
6 Alkane oxidation with alkyl hydroperoxides
Compound 21 catalyses [101] also the alkane oxidation with tert-butyl hydroperoxide. In comparison with the corresponding H2O2 oxidation, the reaction of cyclooctane proceeds much slower and gives lower yields of the products. Another feature of the oxidation with tert-BuOOH is the weaker effect of the acetic acid addition on the efficiency of the catalytic reaction. Indeed, the oxidation with tert-BuOOH in MeCN proceeds also in the absence of MeCO2H. Acetic acid accelerates the oxidation of cyclohexane, the maximum being observed at [MeCO2H] = ca 0.1–0.2 mol dm–3. The kinetic curve shapes and the cyclohexanone/cyclohexanol ratio depend on the acetic acid concentration. The reaction is of first order with respect to the catalyst (compound 21) concentration, because the initial rate of the reaction (measured as the product yields after 5 h) is proportional to [21]0. The total turnover number attains 470.
7 Hydrocarbon oxidation with peroxy acids
Various vanadium complexes (particularly, n-Bu4NVO3) catalyse alkane oxidations by peroxyacetic acid in acetonitrile at 60 °C [102]. The reaction gives a mixture of corresponding ketones, alcohols and alkylacetates; formation of alkyl hydroperoxides can be detected (by reduction with triphenylphosphine; see above) only at the beginning of the reaction. Bond selectivities of the oxidation are not high, which attests the formation of free radicals. Analogous ‘modelling’ reactions with H2O2 in acetonitrile in the presence of acetic acid or in pure acetic acid gave alkyl hydroperoxides as main products. Copper derivative Cu(CH3CN)4BF4, perchlorate and some other complexes of this metal, taken in small concentrations (for example, 10–5 mol dm–3), are also efficient in alkane oxidations with peroxyacetic acid in acetonitrile solution [103]. In this case, the reaction gives rise to the formation of alkyl hydroperoxides as main products and occurs with low bond selectivity. The total turnover number attains 1900. Finally, manganese (IV) complex 21 catalyses alkane oxidations with peroxyacetic or m-chloroperbenzoic acids [85,86].
8 Conclusions
In the last decade, we discovered and investigated a few oxidizing systems based on transition metal complexes as catalysts. The oxygenations distinguish significantly in their efficiencies and selectivities and proceed via different mechanisms. In some cases, these mechanisms involve the formation of free hydroxyl radicals; in other cases, only transient formation of radical-like species can be assumed. Many of these reactions occur only if certain co-catalysts are added in relatively small concentrations. Some of the systems described here are very efficient in oxidations and can be employed in industrial processes.
Acknowledgements
The investigations described in this paper have been carried out partly in cooperation between author’s group and scientific groups of D. Attanasio (Italy), C. Bolm (Germany), J.-M. Brégeault (France), R.S. Drago (USA), Y. Ishii (Japan), E.R. Lachter (Brazil), J.R. Lindsay Smith (UK), D. Mandelli (Brazil), J. Muzart (France), U. Schuchardt (Brazil), G. Süss-Fink (Switzerland), J. Vicente (Spain). I thank the Russian Basic Research Foundation for support.