

1 Introduction
Numerous compounds bearing a polyalkyl-substituted cyclohexane backbone have been developed for their applications in flavour chemistry and perfume industry. Among them are important fragrant compounds such as menthols [1], carvones [2], ionones [3] and damascones [4]. Various substances show a combination of conjugate or non-conjugate double bonds with a carbonyl group in correlation with the natural pattern of the 2,6,6-trimethyl or 1-methyl-4-isopropyl substituents. In the group of ionones, structure–odour relationship has shown that the position of the double bond in the nucleus affects the quality but not the type of odour, while the introduction of a second double bond destroys the violet odour [5]. Furthermore, ionone derivatives having a gem-dimethyl group ortho and a methyl group para to the side chain exhale a closed violet-odour. Among the numerous syntheses of both natural and synthetic cyclohexane derivatives, we took notice of 2,4-dimethyl-cyclohex-3-ene carboxaldehyde 1 [6] as an attractive starting substrate for syntheses of more elaborate derivatives with potential olfactory properties. We have chosen compound 1 as the starting synthon for the following reasons: (a) it possesses olfactory properties; (b) it could lead to analogs of both natural and synthetic olfactive compounds such as α-ionone 2, β-irone 3, 4-(4-methyl-3-penten-1-yl)-3-cyclohexene carboxaldehyde 4 [7] or 4-(2,2,4-trimethyl-cyclohex-3-enyl)-but-3-en-2-one 5 [5b] (Scheme 1); (c) it constitutes a raw material since it is industrially produced by Diels–Alder reaction starting from acrolein and 2-methylpentadiene and is available in a large scale; (d) our aim was to investigate syntheses of compounds with a high added-value including at least one equivalent of acrolein.
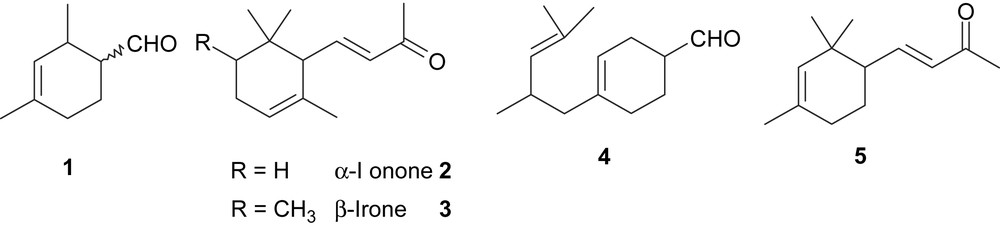
Aldehyde 1 is usually produced in standard conditions of Diels–Alder reaction between acrolein and 2-methylpentadiene as a racemic mixture of cis- and trans-diastereomer (80:20 ratio) besides a small amount of the regioisomer 2,4-dimethylcyclohex-2-ene carboxaldehyde. The reaction can be conducted in the presence of Lewis acid catalysts or metal transition catalysts to improve the stereoselectivity [8,9].
2 Results and discussion
The aim of the study was to synthesize derivatives of compound 1 with functions that are likely to provide a pleasant olfactory note using simple methodology associated, if possible, with low-cost reactives so as to allow possible industrial transfer of the process. First experiments were conducted to synthesize various esters from the acid 6 produced by oxidation of 1 using Ag2O in basic conditions (Scheme 2).
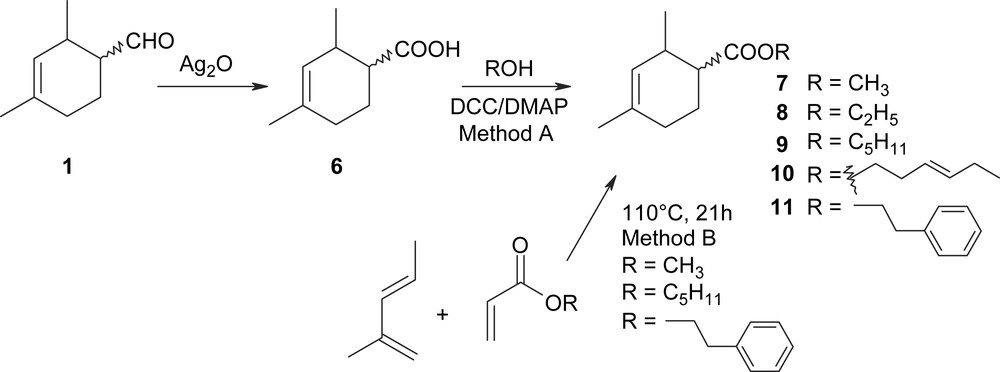
Esterification of 6 in acidic conditions via the acyl chloride or directly by the action of alcohols failed. Under these conditions, we observed the isomerization of the double bond in position 4 and the formation of numerous by-products resulting from the addition of hydrogen chloride or alcohol on the double bond. Finally, the esterification of 6 was performed (method A), at room temperature, in mild conditions using dicyclohexylcarbodiimide (DCC) as the coupling agent in the presence of the alcohol and dimethylaminopyridine (DMAP). Five esters 7–11 were synthesized with moderate yields (55–63%). Among them, the methyl ester 7, the pentyl ester 9 and the phenylethyl ester 11, have revealed interesting olfactory characteristics (see Table 1). Therefore, we searched a cheap alternative procedure. As already reported with ethyl acrylate and various dienes [10], Diels–Alder reaction between methyl acrylate and 2-methylpentadiene led to the corresponding adduct 7 with a good yield (74%). The reaction was conducted at 110 °C for 21 h in the presence of a small amount of hydroquinone to avoid acrylate polymerization (method B). Under the same conditions, pentyl acrylate and phenylethyl acrylate conducted to the adducts 9, 11 in good yields (87 and 83%). The stereoselectivity of the addition was determined by GC–MS analysis of the distillate. As expected, in each case, the major cis/trans-diastereomers were obtained beside a minor regioisomer as described above for the synthesis of 1 (further details are indicated in Section 4).
Olfactory properties of compounds
| Compounds | Quality | Referents [14] |
| 7 | Hesperidic-fruity | Isobutylquinoleine/nootkatone |
| 8 | Fruity | Ethyl isobutyrate/benzyl acetate |
| 9 | Ester-watermelon like | Ethyl isobutyrate/nonanal/decadienal |
| 10 | Ester-fruity/green | Ethyl isobutyrate/nonanal/hexenyl acetate |
| 11 | Soft rosy odour | Ethyl isobutyrate/phenylethylic alcohol |
| 12 | Floral | Benzyl acetate |
| 13 | Floral | Benzyl acetate/naphthaline |
| 14 | Fatty acidic | Isovaleric acid |
| 15 | Green ester/fatty | Cyclopentanone/cis-3-hexenol/nonanal |
| 16 | Aldehydic/camphor | cis-3-Hexenol/camphor |
| 20 | Hesperidic-fruity | Nootkatone |
| 21 | Angelic-type-terpenic ester | Styralyl acetate |
A Wittig reaction between 1 and substituted-methylenetriphenylphosphoranes led to the syntheses of compounds bearing a functionalized unsaturated lateral chain (Scheme 3).

The reaction conducted for 15 h at reflux in the presence of toluene with carbomethoxy or carboethoxymethylenetriphenylphosphorane yields the expected unsaturated esters 12 and 13 (66 and 83%). Similarly and under the same conditions, the formylmethylenetriphenyl-phosphorane led to the unsaturated aldehyde 14 in a poor yield (12%) accompanied with a large amount of the starting compound 1. Other protocols, notably using other solvents, prolonged heating or higher temperature, have failed in all cases. Finally, the reaction of 1 with methoxymethylenetriphenylphosphorane at 0 °C for 30 min in toluene led to the enol ether 15, which was hydrolysed in a mixture of acetic acid–water–THF (3:1:1) at room temperature for 5 days, giving the aldehyde 16, homologue of the starting compound 1. 1H NMR analyses indicated that in the structures 12–14, the ethylenic protons display coupling constants characteristic of an E double bond. On the other hand, the NMR pattern of compound 15 supported the presence of a mixture of Z and E stereoisomers (57:43 ratio).
Then, according to the aim of our study, we focused our interest on the Grignard reagent derived from 2-(2-bromoethyl)-[1,3]dioxane 17, which is directly derived from acrolein. As indicated in Scheme 4, a Grignard reaction between 1 and the bromo derivative 17, conducted in the conditions described elsewhere [11], gave the alcohol 18 in a very good yield (91%).
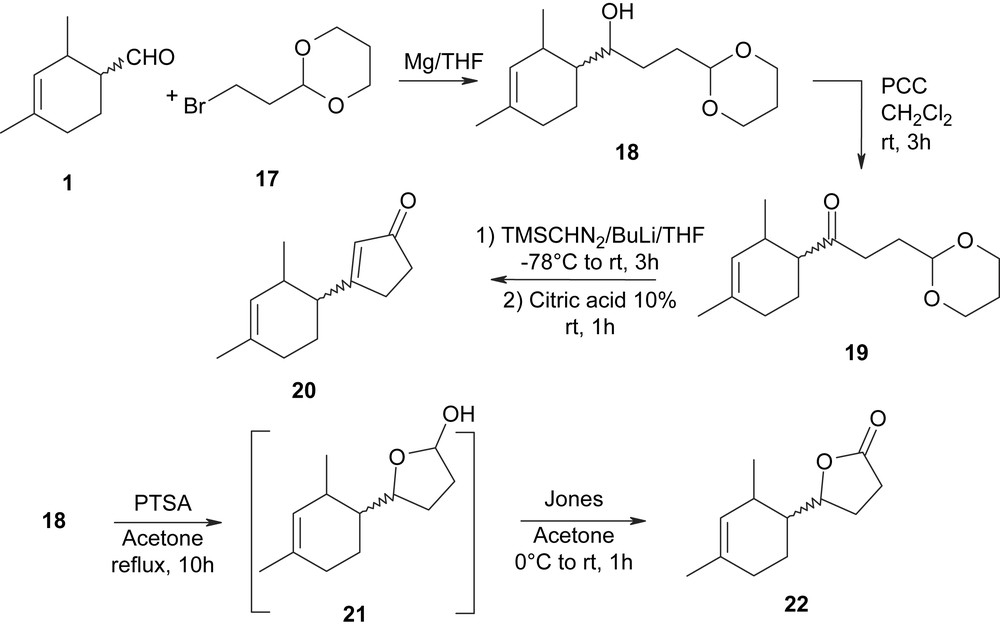
Oxidation of the alcohol 18 using pyridinium chlorochromate [12] in methylene chloride at room temperature for 3 h led to the ketone 19 in good yield (84%). The ketone 19 was then reacted with lithium trimethylsilyldiazomethane [13] to give, after hydrolysis in diluted citric acid of the intermediate acetal which was not isolated, the cyclopentenone 20 in a moderate yield (40%). Finally, treatment of the alcohol 18 with para-toluene sulfonic acid at reflux in a mixture of acetone–water (3:1) gave the lactol 21. This latter was not isolated, but oxidized using the Jones reagent in acetone at 0 °C for 1 h, leading to the lactone 22 in a moderate yield (56%).
3 Olfactory evaluations
Table 1 summarizes the olfactive properties of compounds 7–16 and 20, 22.
Starting aldehyde 1 exhibits an extremely strong accentuated grassy-green note with a herbal camphor side note. Esters 7 and 8 are hesperidic-fruity odorants with grapefruit notes. Compound 9 possesses typical ester notes with an aldehydic tonality and a watermelon odour which is currently attractive and could well be applied to perfume compositions. Ester 10 is fruity-green with a fatty aldehydic character. In contrast, compound 11 is not very powerful, possessing a rosy odour weaker than phenylethylic alcohol. Esters 12 and 13 which could be considered as vinylic analogs of esters 7 and 8 show, in contrast, a heavy floral note. Compound 12 shows a linear odour with a good tenacity while the floral odour of compound 13 is less interesting than ester 12 because of an unpleasant aromatic undertone. In contrast to compound 1, aldehyde 14 is characterized by an unpleasant fatty acidic odour similar to isovaleric acid. Olfactory properties of 15 and 16 are interesting since they show two facets. Compound 16 exhibits a green ester and fatty note, while aldehyde 16 shows a typical aldehydic note associated with a terpenic by-note. Acetals 18 and 19 do not present any significative odour. Finally, cyclopentenone 20 possesses a hesperidic-fruity note like nootkatone, but shows weaker tenacity, and the lactone 21 has terpenic odorant with an angelic-type odour like styralyle acetate.
The odour evaluations performed on our samples pointed out that all these compounds had very different odour properties in spite of their common dimethylcyclohexenyl skeleton. Compounds 7, 9, 15 and 16 which were selected by the olfactory evaluation panel to possess interesting fragrance could be used in flowery or fruity compositions.
4 Experimental section
4.1 General procedure
Analytical thin layer chromatography was performed on precoated plates of silica gel 60F 254 (Merck) and column chromatography on Nacherey Nagel silica gel (230–400 mesh). GC analyses were carried out on GC/MS Hewlett Packard 5890A, detector HP5970, column supelco SPB-1 (polydimethylsiloxane). The infrared spectra were recorded on a Perkin–Elmer FTIR paragon 1000 spectrometer. 1H and 13C NMR spectra were recorded on a Brucker AC 200 instruments using CDCl3 as solvent, and chemical shifts (δ) are expressed in parts per million relative to residual CHCl3 at δ = 7.27 for 1H. All the compounds described herein are obtained as a mixture of diastereoisomers. For each compound we indicated the 1H NMR data of the major and minor diastereoisomers, except for compound 10 which was obtained as a mixture of two diastereoisomers in a proportion not determined by NMR (determined by GC) and compound 15, which was obtained as a mixture of 4 identified diastereoisomers. In all cases, the 13C NMR spectra are in accordance with the structures.
4.2 2,4-Dimethylcyclohex-3-ene carboxylic acid (6)
An aqueous solution of 1 (20 g, 0.14 mol), Ag2O (34.8 g, 0.15 mol) and NaOH (27 g, 0.67 mol) was stirred at room temperature for 15 h and then diluted with diethyl ether (100 ml). The aqueous layer was slowly acidified to pH = 2 with 10% aqueous HCl and extracted with diethyl ether. The organic phase was dried over anhydrous MgSO4, filtered and evaporated. The crude product (15.9 g, 74% yield) which was obtained as a mixture of two diastereoisomers (80:20 ratio, stereochemistry not determined) was pure enough to be used without further purification in the next step.
mp = 91 °C (lit. mp = 92–93 °C [9]). IR (film) ν (cm−1): 1702 (CO). 1H NMR δ (ppm): 0.92 (d, J = 7.0 Hz, 3H, minor diastereoisomer, 20%), 0.98 (d, J = 7.0 Hz, 3H, major diastereoisomer, 80%), 1.55–2.12 (m, 4H), 1.65 (s, 3H), 2.42–2.48 (m, 1H), 2.50–2.72 (m, 1H), 5.17 (s, 1H, major diastereoisomer, 80%), 5.30–5.39 (m, 1H, minor diastereoisomer, 20%).
4.3 General procedure for preparation of esters 7–11 (method A)
A solution of 6 (3 g, 19.4 mmol), alcohol (23 mmol), dicyclohexylcarbodiimide (DCC) (4.42 g, 21.3 mmol), and dimethylaminopyridine (0.2 g, 1.64 mmol) in dichloromethane (30 ml) was stirred at room temperature for one day under inert conditions. The excess of DCC was hydrolysed with 10% aqueous HCl (3 ml). The mixture was stirred for 1 h and extracted with diethyl ether (2 × 50 ml). The organic layer was washed with water (3 × 20 ml), dried over anhydrous MgSO4, filtered and evaporated. The crude product was purified by chromatography on silica gel (hexane–ethylacetate 85:15 v/v).
4.3.1 Methyl(2,4-dimethyl-cyclohex-3-ene)carboxylate (7)
This compound was obtained using methanol with 59% yield as a colorless liquid and as a mixture of two diastereoisomers (65:35 ratio, stereochemistry not determined).
IR (film) ν (cm−1): 1738 (CO). 1H NMR δ (ppm): 0.82 (d, J = 7.0 Hz, 3H, minor diastereoisomer, 35%), 0.91 (d, J = 7.0 Hz, 3H, major diastereoisomer, 65%), 1.52–2.12 (m, 4H), 1.62 (s, 3H), 2.18–2.60 (m, 2H), 3.64 (s, 3H), 5.12 (s, 1H, major diastereoisomer, 65%), 5.32 (s, 1H, minor diastereoisomer, 35%). MS (EI): m/z 168 (M+, 15), 137 (7), 136 (7), 125 (5), 121 (4), 109 (41), 108 (100), 93 (92), 77 (18), 67 (47), 55 (19).
4.3.2 Ethyl(2,4-dimethyl-cyclohex-3-ene)carboxylate (8)
This compound was obtained using ethanol with 59% yield as a colorless liquid and as a mixture of two diastereoisomers (55:45 ratio, stereochemistry not determined).
IR (film) ν (cm−1): 1731 (CO). 1H NMR δ (ppm): 0.83 (d, J = 7.0 Hz, 3H, minor diastereoisomer, 45%), 0.93 (d, J = 7.0 Hz, 3H, major diastereoisomer, 55%), 1.23 (t, J = 7.5 Hz, 3H), 1.55–2.22 (m, 4H), 1.62 (s, 3H), 2.25–2.68 (m, 2H), 4.11 (q, J = 7 Hz, 2H, minor diastereoisomer, 45%), 4.12 (q, J = 7 Hz, 2H, major diastereoisomer, 55%), 5.15 (s, 1H, minor diastereoisomer, 45%), 5.25 (s, 1H, major diastereoisomer, 55%). MS (EI): m/z 182 (M+, 11), 153 (6), 137 (10), 125 (5), 121 (4), 109 (43), 108 (100), 107 (41), 93 (57), 91 (15), 79 (15), 77 (15), 67 (32), 55 (15).
4.3.3 Pentyl(2,4-dimethyl-cyclohex-3-ene)carboxylate (9)
This compound was obtained using pentyl alcohol with 63% yield as a colorless liquid and as a mixture of two diastereoisomers (55:45 ratio, stereochemistry not determined).
IR (film) ν (cm−1): 1732 (CO). 1H NMR δ (ppm): 0.80–0.97 (m, 6H), 1.18–1.44 (m, 2H), 1.50–2.10 (m, 8H), 1.65 (s, 3H), 2.30–2.60 (m, 2H), 3.75–4.10 (m, 2H), 5.15 (m, 1H, minor diastereoisomer, 45%), 5.31 (m, 1H, major diastereoisomer, 55%). MS (EI): m/z 224 (M+, 13), 153 (19), 137 (10), 136 (15), 109 (41), 108 (100), 93 (28), 67 (22), 55 (16).
4.3.4 cis-3-Hex-3-enyl(2,4-dimethyl-cyclohex-3-ene)carboxylate (10)
This compound was obtained using cis-3-hex-3-enyl alcohol with 55% yield as a colorless liquid and as a mixture of two diastereoisomers (55:45 ratio, determined by gas chromatography).
IR (film) ν (cm−1): 1733 (CO). 1H NMR δ (ppm): 0.79–1.01 (m, 6H), 1.55–2.18 (m, 8H), 1.58 (s, 3H), 2.25–2.60 (m, 2H), 3.92–4.11 (m, 2H), 5.12–5.55 (m, 3H). MS (EI): m/z 236 (M+, 2), 154 (15), 153 (36), 136 (8), 109 (35), 108 (47), 107 (100), 93 (30), 83 (38), 67 (53), 55 (95).
4.3.5 Phenethyl(2,4-dimethyl-cyclohex-3-ene)carboxylate (11)
This compound was obtained using 1-phenylethyl alcohol with 55% yield as a colorless liquid and as a mixture of two diastereoisomers (51:49 ratio, stereochemistry not determined).
IR (film) ν (cm−1): 1732 (CO). 1H NMR δ (ppm): 0.76 (d, J = 7 Hz, 3H, minor stereoisomer, 49%), 0.90 (d, J = 7 Hz, 3H, major diastereoisomer, 51%), 1.58–2.22 (m, 4H), 1.63 (s, 3H), 2.25–2.56 (m, 2H), 2.95 (t, J = 7.0 Hz, 2H), 4.23–4.38 (m, 2H), 5.18 (m, 1H, minor diastereoisomer, 49%), 5.35 (m, 1H, major diastereoisomer, 51%), 7.12–7.55 (m, 5H). MS (EI): m/z 258 (M+, 7), 153 (25), 108 (22), 107 (31), 105 (100), 91 (19), 79 (20), 67 (16).
4.4 General procedure for preparation of esters 7, 9 and 11 (method B)
A mixture of 2-methyl-1,3-pentadiene (6.9 g, 84 mmol), hydroquinone (0.1 g, 0.084 mmol) and methyl-, pentyl- or phenylethyl acrylate (70 mmol) was stirred at 120 °C for one day in a reactor. After cooling, distillation of the crude product afforded the expected esters as a mixture of two diastereoisomers (75:25).
4.5 Methyl[3-(2,4-dimethyl-cyclohex-3-enyl)]acrylate (12)
A solution of 1 (3 g, 22.7 mmol) in toluene (30 ml) was slowly added to a solution of carbomethoxymethylenetriphenylphosphorane (7.8 g, 22.7 mmol) in toluene (30 ml) under inert conditions. The solution was refluxed for 15 h and the solvent was removed under reduced pressure. The residue was diluted with diethylic ether (40 ml) and the solution was stirred for 1 h. The solution was filtered and insoluble triphenylphosphine was washed with diethyl ether (3 × 20 ml). The organic extracts were collected, dried over MgSO4, filtered and concentrated. Purification of the residue by silica gel chromatography (hexane–ethylacetate 80:20) afforded 2.9 g (66% yield) of 12 as a colorless liquid and as a mixture of two diastereoisomers (70:30 ratio, stereochemistry not determined).
IR (film) ν (cm−1): 1726 (CO), 1652 (CC). 1H NMR δ (ppm): 0.82 (d, J = 7 Hz, 3H, major diastereoisomer, 70%), 0.92 (d, J = 7 Hz, 3H, minor diastereoisomer, 30%), 1.63–1.87 (m, 2H), 1.65 (s, 3H), 1.89–2.12 (m, 2H), 2.21–2.57 (m, 2H), 3.70 (s, 3H), 5.18 (s, 1H, minor diastereoisomer, 30%), 5.28 (s, 1H, major diastereoisomer, 70%), 5.82 (dd, J = 1.7 Hz, J = 16.1 Hz, 1H), 6.87 (dd, J = 8.6 Hz, J = 15.6 Hz, 1H, minor diastereoisomer, 30%), 6.98 (dd, J = 8.1 Hz, J = 15.6 Hz, 1H, major diastereoisomer, 70%). MS (EI): m/z 194 (M+, 10), 135 (7), 108 (11), 93 (10), 82 (100), 67 (64), 53 (15).
4.6 Ethyl[3-(2,4-dimethyl-cyclohex-3-enyl)]acrylate (13)
A procedure similar to that described above for preparation of compound 12 afforded 4.1 g (88% yield) of 13 as a colorless liquid and as a mixture of two diastereoisomers (75:25 ratio, stereochemistry not determined).
IR (film) ν (cm−1): 1718 (CO), 1654 (CC). 1H NMR δ (ppm): 0.80 (d, J = 7.4 Hz, 3H, major diastereoisomer, 75%), 0.90 (d, J = 7.4 Hz, 3H, minor diastereoisomer, 25%), 1.25 (t, J = 7 Hz, 3H) 1.58–1.80 (m, 2H), 1.61 (s, 3H), 1.89–2.20 (m, 2H), 2.22–2.55 (m, 2H), 4.13 (q, J = 7.5 Hz, 2H), 5.16 (s, 1H, minor diastereoisomer, 25%), 5.24 (s, 1H, major diastereoisomer, 75%), 5.82 (dd, J = 1.6 Hz, J = 15.6 Hz, 1H), 6.86 (dd, J = 8.1 Hz, J = 15.6 Hz, 1H, minor diastereoisomer, 25%), 6.98 (dd, J = 8.1 Hz, J = 15.6 Hz, 1H, major diastereoisomer, 75%). MS (EI): m/z 208 (M+, 8), 135 (8), 108 (10), 82 (100), 67 (53), 53 (12).
4.7 3-(2,4-Dimethyl-cyclohex-3-enyl)-propenal (14)
A mixture of 1 (9 g, 6.51 mmol) and triphenylphosphoranylidene acetaldehyde (2 g, 6.57 mmol) in toluene (100 ml) was refluxed for 9 h under inert atmosphere. The mixture was concentrated and the residue was purified by chromatography on silica gel (cyclohexane–dichloromethane, 70:30) to give 14 (1.28 g, 12% yield) as a colorless liquid and as a mixture of two diastereoisomers (60:40 ratio, stereochemistry not determined).
IR (film) ν (cm−1): 1686 (CO), 1633 (CC). 1H NMR δ (ppm): 0.85 (d, J = 7.1 Hz, 3H, major diastereoisomer, 60%), 0.93 (d, J = 7.1 Hz, 3H, minor diastereoisomer, 40%), 1.25 (t, J = 7 Hz, 1H) 1.45–2.10 (m, 4H), 1.55 (s, 3H), 2.10–2.72 (m, 2H), 5.20 (s, 1H, minor diastereoisomer, 40%), 5.38 (s, 1H, major diastereoisomer, 60%), 6.12 (dd, J = 7 Hz, J = 15.6 Hz, 1H), 6.76 (dd, J = 7.8 Hz, J = 15.6 Hz, 1H, minor diastereoisomer, 40%), 6.87 (dd, J = 7.8 Hz, J = 15.6 Hz, 1H, major diastereoisomer, 60%), 9.49 (d, J = 7.8 Hz, 1H). MS (EI): m/z 164 (M+, 5), 149 (12), 108 (18), 107 (16), 92 (18), 82 (85), 67 (100), 53 (20).
4.8 4-(2-Methoxy-vinyl)-1,3-dimethyl-cyclohex-3-ene (15)
A solution of LDA in THF was prepared as follows: to a solution of THF (50 ml) and distilled diisopropylamine (1.2 ml, 8.6 mmol) was added dropwise a solution of n-butyllithium 2.5 M (3.5 ml, 8.7 mmol) at −30 °C under inert atmosphere. The mixture was stirred at this temperature for 30 min and was allowed to react for an additional 30 min at room temperature. The solution of LDA was added dropwise to a mixture of methoxymethyltriphenylphosphonium chloride (2.8 g, 8.2 mmol) in toluene (10 ml) at 0 °C under inert atmosphere. After stirring for 30 min, a solution of 1 (1 g, 7.2 mmol) in toluene (5 ml) was added and the mixture was allowed to react at 0 °C for 30 min and then hydrolysed successively with a saturated aqueous NH4Cl solution (7 ml) and with water (10 ml) to dissolve precipitated salts. The solution was extracted with diethylic ether (3 × 5 ml) and the organic phase was washed with brine (2 × 5 ml). After drying over anhydrous MgSO4, the solvent was evaporated under reduced pressure and the residue was purified by chromatography on silica gel (hexane–ethylacetate, 95:5) to give 15 (0.67 g, 56% yield) as a colorless liquid and as a mixture of four diastereoisomers Z-cis, Z-trans, E-cis and E-trans (47:10:30:13 ratio, respectively).
1H NMR δ (ppm): 0.78–0.94 (m, 3H), 1.52–2.02 (m, 4H), 1.61 (s, 3H), 2.10–2.38 (m, 7H), 2.72–2.88 (m, 1H), 3.52 (s, 3H, E diastereoisomers, 43%), 3.57 (s, 3H, Z diastereoisomers, 57%), 4.18 (dd, J = 6.5 Hz, J = 9.7 Hz, 1H, trans diastereoisomers, 23%), 4.30 (dd, J = 6.5 Hz, J = 9.7 Hz, 1H, cis diastereoisomers, 77%), 4.58 (dd, J = 8.1 Hz, J = 12.9 Hz, 1H, trans diastereoisomers, 23%), 4.59 (dd, J = 9.1 Hz, J = 13 Hz, 1H, cis diastereoisomers, 77%), 5.12–5.28 (m, 1H), 5.78 (dd, J = 1.1 Hz, J = 6.5 Hz, 1H, Z diastereoisomers, 57%), 6.30 (d, J = 12.4 Hz, 1H, E diastereoisomers, 43%). MS (EI): m/z 166 (M+, 2), 108 (14), 84 (100), 69 (28).
4.9 (2,4-Dimethyl-cyclohex-3-enyl)-acetaldehyde (16)
A solution of compound 15 (0.5 g, 3 mmol) in 3 ml of a mixture of acetic acid, THF and water (3:1:1) was stirred under inert atmosphere for 5 days. The solvent was evaporated under reduced pressure and the residue was purified by chromatography on silica gel (hexane–ethylacetate, 80:20) to give 16 (0.2 g, 44% yield) as a colorless liquid and as a mixture of two diastereoisomers (70:30 ratio, stereochemistry not determined).
IR (film) ν (cm−1): 1725 (CO). 1H NMR δ (ppm): 0.82 (d, J = 7.0 Hz, 3H, minor diastereoisomer, 30%), 0.96 (d, J = 7 Hz, 3H, major diastereoisomer, 70%), 1.15–2.42 (m, 7H), 1.60 (s, 3H), 2.52 (ddd, J = 1.6 Hz, J = 4.3 Hz, J = 16.1 Hz, 1H), 5.16 (s, 1H, major diastereoisomer, 70%), 5.25 (s, 1H, minor diastereoisomer, 30%), 9.74 (t, J = 1.6 Hz, 1H). MS (EI): m/z 152 (M+, 2), 108 (100), 93 (95), 91 (21), 82 (23), 77 (20), 67 (66), 55 (18).
4.10 1-(2,4-Dimethyl-cyclohex-3-enyl)-3-[1,3]dioxan-2-yl-propan-1-ol (18)
To a suspension of magnesium (0.3 g, 12.3 mmol) in anhydrous THF (4 ml) was added dropwise a solution of 17 (2.1 g, 10.8 mmol) in anhydrous THF (15 ml). The temperature was kept at 35 °C during the addition of the halide and the mixture was allowed to react for 30 min at room temperature. A solution of 1 (1 g, 7.2 mmol) in anhydrous THF (6 ml) was then added dropwise at room temperature and the mixture was allowed to react again for 15 min. The reaction was carefully quenched with saturated aqueous NH4Cl solution (2 ml) and diethylic ether (8 ml) was then added. The precipitate was filtered and washed with diethylic ether. The organic phases were collected, washed with water (3 × 4 ml), dried over anhydrous MgSO4 and filtered. Evaporation of the solvent and recrystallization of the residue from ethanol–water (40:60) led to compound 18 (1.67 g, 91% yield) as a white solid and as a mixture of two diastereoisomers (75:25 ratio, stereochemistry not determined).
mp = 92 °C. IR (film) ν (cm−1): 3017 (OH). 1H NMR δ (ppm): 0.75 (d, J = 7 Hz, 3H, major diastereoisomer, 75%), 0.88 (d, J = 7 Hz, 3H, minor diastereoisomer, 25%), 1.15–2.25 (m, 8H), 1.59 (s, 3H), 1.75 (s, 2H), 1.89 (s, 2H), 2.35 (d, J = 4.7 Hz, 1H), 3.30–3.48 (m, 1H), 3.72 (t, J = 11 Hz, 2H), 4.05 (dd, J = 4.7 Hz, J = 11 Hz, 2H), 4.41 (t, J = 4.7 Hz, 1H, minor diastereoisomer, 25%), 4.51 (t, J = 4.7 Hz, 1H, major diastereoisomer, 75%), 5.14 (d, J = 4.4 Hz, 1H, minor diastereoisomer, 25%), 5.26 (d, J = 4.4 Hz, 1H, major diastereoisomer, 75%). MS (EI): m/z 253 (M+, 2), 178 (35), 145 (48), 106 (42), 87 (100), 71 (55), 67 (54).
4.11 1-(2,4-Dimethyl-cyclohex-3-enyl)-3-[1,3]dioxan-2-yl-propan-1-one (19)
A solution of 18 (3 g, 11.8 mmol) in CH2Cl2 (45 ml) was added dropwise to a solution of pyridinium chlorochromate (6.4 g, 29.7 mmol) in CH2Cl2 (45 ml) at room temperature. After 3 h reaction at room temperature, the solvent was eliminated under reduced pressure and the residue was diluted with diethylic ether (100 ml). The organic phase was successively washed with saturated NaHCO3 solution (40 ml), water (2 × 20 ml), dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by chromatography on silica gel (cyclohexane–diethylic ether, 80:20) to give 19 (2.5 g, 84% yield) as a white solid and as a mixture of two diastereoisomers (95:5 ratio, stereochemistry not determined).
mp = 31 °C. IR (film) ν (cm−1): 1710 (CO), 1377 (C–O–C). 1H NMR δ (ppm): 0.72 (d, J = 6.3 Hz, 3H, major stereoisomer, 95%), 0.82 (d, J = 6.3 Hz, 3H, minor stereoisomer, 5%), 1.30 (d, J = 13.3 Hz, 2H), 1.52–2.18 (m, 6H), 1.61 (s, 3H), 2.34–2.72 (m, 4H), 3.70 (t, J = 11.4 Hz, 2H), 3.90 (dd, J = 3.9 Hz, J = 11 Hz, 2H), 4.48 (t, J = 5.5 Hz, 1H), 5.35 (s, 1H). MS (EI): m/z 252 (M+, 5), 251 (5), 176 (55), 143 (22), 132 (25), 109 (60), 100 (85), 87 (58), 85 (100), 67 (58).
4.12 3-(2,4-Dimethyl-cyclohex-3-enyl)-cyclopent-2-enone (20)
A 2.2 M solution of n-butyllithium in hexane (0.6 ml, 1.32 mmol) was added to a 2 M solution of trimethylsilyldiazomethane (0.6 ml, 1.2 mmol) in hexane diluted with anhydrous THF (2 ml) at −78 °C under inert conditions. After 1 h of reaction, a solution of 19 (0.23 g, 0.91 mmol) in THF (5 ml) was added dropwise at the same temperature. The mixture was allowed to react at −78 °C for 1 h and allowed to reach room temperature over a period of 1 h. The solution was acidified with 10% citric acid solution (10 ml) under stirring for 1 h and extracted with ethylacetate (15 ml). The organic phase was washed with saturated NaCl solution (7 × 5 ml), dried over MgSO4, filtered and concentrated. The residue was purified by chromatography on silica gel (cyclohexane–ethylacetate, 90:10) to give 20 (70 mg, 40% yield) as a colorless liquid and as a mixture of two diastereoisomers (70:30 ratio, stereochemistry not determined).
IR (film) ν (cm−1): 1711 (CO), 1679 and 1609 (CC). 1H NMR δ (ppm): 0.75 (d, J = 7.1 Hz, 3H, major diastereoisomer, 70%), 0.84 (d, J = 7.1 Hz, 3H, minor diastereoisomer, 30%), 1.52–1.85 (m, 2H), 1.62 (s, 3H), 1.86–2.31 (m, 2H), 2.35–2.75 (m, 6H), 5.21 (s, 1H, minor diastereoisomer, 30%), 5.40 (s, 1H, major diastereoisomer, 70%), 5.90 (d, J = 1.57 Hz, 1H, major diastereoisomer, 70%), 5.95 (d, J = 1.57 Hz, 1H, minor diastereoisomer, 30%). MS (EI): m/z 190 (M+, 35), 109 (75), 82 (100), 67 (90).
4.13 5-(2,4-Dimethyl-cyclohex-3-enyl)-3-[1,3]dihydro-furan-2-one (22)
A solution of 18 (1.5 g, 5.9 mmol) and para-toluene sulfonic acid (0.15 g, 0.79 mmol) in acetone (45 ml) and water (22.5 ml) was heated at reflux for 10 h. After addition of saturated NaHCO3 solution (2 ml), the mixture was extracted with ether (4 × 15 ml) and the organic phases were washed with water (2 × 10 ml), dried over anhydrous MgSO4, filtered and concentrated. The residue was dissolved in acetone (50 ml), cooled at 0 °C, and Jones reagent (2.2 ml, 5.9 mmol) was added dropwise over a period of 30 min. The reaction mixture was allowed to reach room temperature and was hydrolysed with water (100 ml). After extraction with diethylic ether (3 × 100 ml), the collected organic phases were washed with 5% NaHCO3 solution, dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by distillation under reduced pressure (Eb3 mm Hg = 140 °C) to give 22 as a white solid which slowly crystallizes (0.65 g, 56% yield) and as a mixture of two diastereoisomers (95:5 ratio, stereochemistry not determined).
mp = 40 °C. IR (film) ν (cm−1): 1770 (CO), 1377 (C–O–C). 1H NMR δ (ppm): 0.85 (d, J = 7.1 Hz, 3H, major stereoisomer, 95%), 0.92 (d, J = 7.1 Hz, 3H, minor stereoisomer, 5%), 1.15–2.10 (m, 6H), 1.68 (s, 3H), 2.12–2.40 (m, 2H), 2.55 (dd, J = 7.1 Hz, J = 10.2 Hz, 2H), 4.30 (ddd, J = 5.5 Hz, J = 8.6 Hz, J = 14.9 Hz, 1H), 5.28–5.39 (m, 1H). MS (EI): m/z 194 (25), 109 (50), 107 (52), 85 (100), 67 (60).
Acknowledgements
Patricia Monnier-Benoit was financially supported by a grant from Agence Nationale de la Recherche Technique and from PCAS SA (Longjumeau, France), a chemical company focused on several expanding markets in fine and specialty chemicals.