

1 Introduction
Nature provides us with most of the finest flavours and fragrances. For decades men have tried to synthesise by themselves these aromatic compounds to fully supply their own needs. Most of these compounds are chiral and some of them show different odour properties according to their absolute configuration [1]. The cheapest and most direct synthetic routes were those aimed to the preparation of racemates. However, there are remarkable examples of enantioselectivity in odour perception, which made compulsory the synthesis of the sole odour active isomer of a certain odorant. Menthol is an outstanding example.
(–)-l-Menthol (Fig. 1) is one of the most important flavours, with an estimated global market of 5000 tons per year. It is used extensively in pharmaceuticals, cosmetics, toothpastes, and chewing gums. The majority of (–)-menthol, ca. 1800–2300 tons per year, is still obtained by freezing the oil of Mentha arvensis (from China) to promote the crystallisation of the menthol contained. This natural menthol is then physically separated by centrifugation and subsequent removal of the supernatant liquid, the so-called dementholized corn-mint oil. Residual impurities are due to traces of Mentha arvensis oil, and may impart a slight peppermint aroma to the menthol crystals. (–)-Menthol is described as very cooling, fresh, sweet and minty, whereas the enantiomer is fresh, only some cooling, sweet-minty with musty, bitter, phenolic and herbaceous notes [2]. Great effort has been devoted to the production of the (–)-enantiomer by synthetic routes starting from readily available raw materials. The two major commercial synthetic sources of (–)-menthol are the Haarman–Reimer (1500–2000 tons per year) and Takasago (1200 tons per year) processes [3, 4].

The first one is based on the fractional crystallisation of menthyl benzoate enantiomers promoted by crystal seeds of optically active menthyl benzoate. m-Cresol and propylene are employed as inexpensive starting materials. The process involves several recycles and leads to a 90% overall yield (Fig. 2).
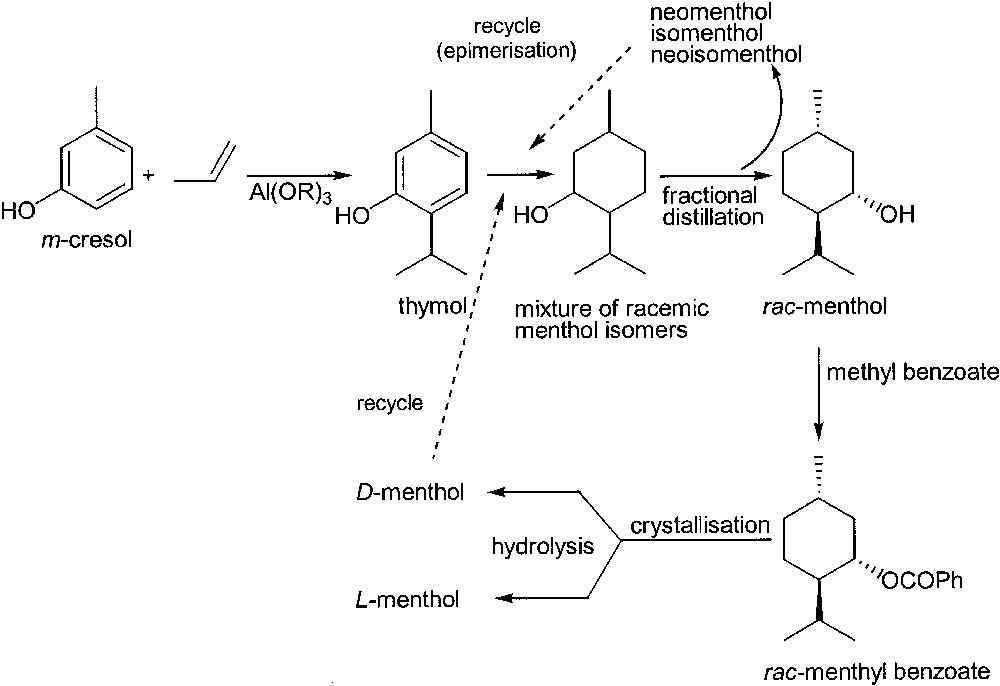
Haarmann and Reimer process for the synthesis of (–)-l-menthol.
In the early 1980s, Takasago developed an elegant synthesis of (–)-menthol from myrcene (Fig. 3). Lithium catalysed addition of diethylamine promoted the conversion of myrcene into diethylgeranylamine. This latter is then isomerised to the chiral (3R)-citronellal N,N-diethyl enamine with 96–99% enantiomeric excess in the presence of RhI-(S)-binap, with a turnover of more than 400 000. Hydrolysis affords (3R)-citronellal of higher optical purity than the one recovered from citronella oil. The conversion of (3R)-citronellal into (–)-menthol is performed through a conventional route.
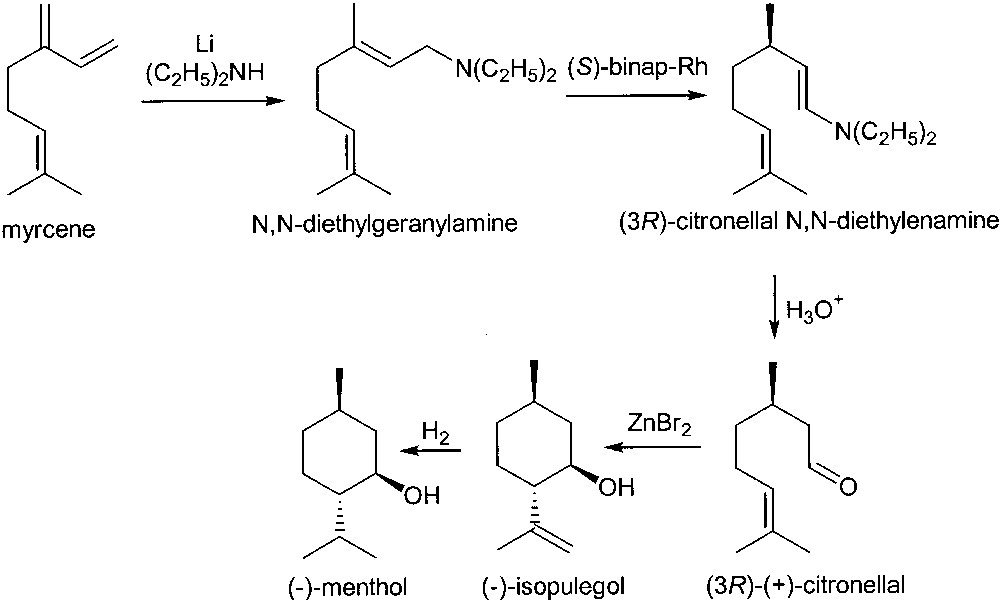
Takasago process for the synthesis of (–)-menthol.
Somewhat different reasons led to the synthesis of the (–)-enantiomer of Ambrox® (Fig. 1). In the realm of fragrances, a relevant role is played by ambergris odorants. Ambergris is produced in the gut of the sperm whale, Physester catodon L. It is usually associated with the beaks of the whale’s principal food, the common cuttlefish. It consists of 80% ambrein, a cholesterol derivative that is considered to be a secretion of the whale’s gut in response to the constant irritation caused by the sharp beaks of the squid. In the gut of the whale, the ambergris appears as a black, viscous and foul-smelling liquid. On exposure to sun light and air, it quickly oxidises and hardens to give a pleasantly aromatic, marbled, greyish, waxy substance in which the squid beaks are still embedded. When it is warmed it produces a very pleasant, mild, sweet, earthy aroma. From ancient times it has been used as a fixative for rare perfumes since it has the effect of making other fragrances last much longer than they would otherwise.
Since the decline of whaling and the restriction of global whaling operations, ambergris has not been used for some time. With the loss of this material, the perfume industry turned to organic synthesis to prepare one of the most appreciated ambergris odorant: (–)-Ambrox® (Fig. 1).
This latter is characterised by a persistent, animalic, amber odour, with an odour threshold of 0.3 ppb. The (+)-enantiomer with its higher threshold value (2.4 ppb vs 0.3 ppb) and accentuated woody note was found to be lacking the strong and warm animal note of the (–)-enantiomer [5], and was, therefore, called “poor man’s Ambrox” by Firmenich perfumers.
(–)-Ambrox® has a complex structure with four stereogenic carbon atoms. It seemed convenient to look for a starting material of natural origin that was an advanced enantiopure intermediate of the whole synthesis. Thus, the most efficient and cheaper access to Ambrox® was found to be the oxidative degradation of (–)-sclareol [6], an abundant constituent of clary sage oil (Salvia sclarea L.) (Fig. 4). Oxidation of the side chain gives γ-lactones (+)-sclareolide (1a) and (–)-iso-sclareolide (1b), which are reduced to a mixture of diastereoisomeric diols 2a and 2b. Cyclisation of these latter derivatives affords (–)-Ambrox and its 8-epi-isomer (3).
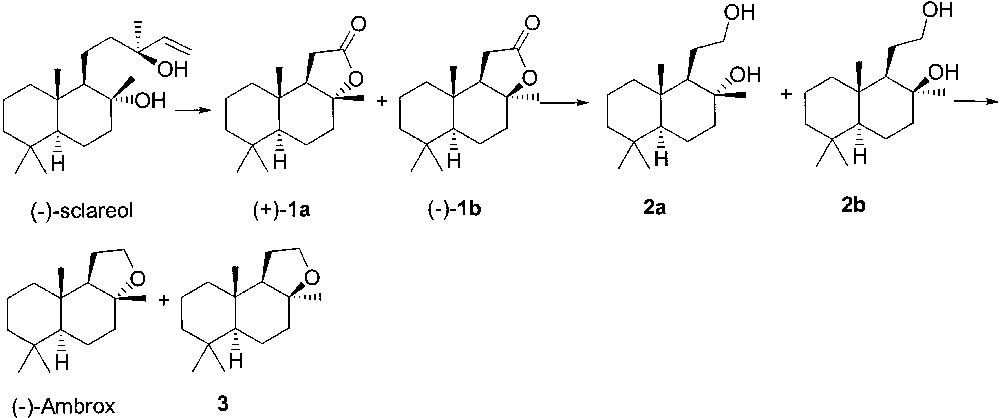
Synthesis of (–)-Ambrox from (–)-sclareol.
Another great challenge was addressed to organic chemists by irones. In the perfumer’s palette, Iris absolute is one of the most expensive products and it is mainly used in luxury perfumes, such as Chanel No. 19 (1970) and So pretty by Cartier (1995). Just to give an insight of the worldwide resonance reached by iris cultivation in the 20th century, we have to take notice that the first multivariate discriminant analysis was performed by R.A. Fisher in 1936 [7] by elaboration of the so called Edgar Anderson’s Iris Data [8]. This latter was a set of measurements of four flower parameters (sepal length and width, petal length and width) of 50 plants from each of the three species of iris: Iris setosa, versicolor, and virginica. The multivariate analysis of Fisher allowed the classification of iris species according to the values of these flower parameters. A taxonomic problem related to a practical interest stimulated the optimisation of a procedure, which is nowadays fundamental for data analysis in modern environmental and food science.
Irones 4–6 (Fig. 5) are the odoriferous principle of iris oil. It is well known that freshly harvested iris rhizomes do not contain irones, but their triterpenoid precursors called ‘iridals’ [9–12]. According to the traditional procedure, decorticated rhizomes are kept in a dry and aerated environment for 2–3 years, then powdered, incubated with diluted sulphuric acid, then steam-distilled to provide the precious ‘Orris butter’. The mechanism of the oxidative degradation affording irones from iridals is still unknown. The traditional process is long, troublesome and low yielding; hence the high cost of the essence (butter). Purification of the essence eliminates the fatty acids and yields the absolute, which is sold at 40 000–50 000 € per kilogram [13].

The odoriferous principle of iris root oil was first isolated by chance by Ferdinand Tiemann, who had started a research in cooperation with Haarmann and Reimer and Laire and co-workers to investigate the components of violet flower oil (Viola odorata L., fam. Violaceae). On the assumption that the two oils contained the same odour vector, the more readily available iris root oil was employed for these extensive investigations. In 1893 [14], Tiemann and Krüger isolated via a phenylhydrazone route a ketonic compound to which they assigned the name irone, and the wrong molecular formula C13H20O. They thought that irone could be the condensation product of citral with acetone. They performed the reaction in laboratory: base-catalysed condensation of citral with acetone gave a compound, they called pseudoionone, which showed a strange, but not characteristic odour. However, when pseudoionone was treated with sulphuric acid in water and glycerine, a ketonic compound was obtained, showing the typical scent of violets in bloom. They called it ionone. They concluded that the isolated irone had to be a double-bond isomer of the synthesised ionone.
In 1933, Ruzicka [15] determined the correct elemental analysis of irone (C14H22O); then he [16] and Naves [17], independently, found that at least three isomers of irone were present in natural iris oil, their structure being determined as 4, 5 and 6 (Fig. 5). In 1971, Rautenstrauch and Ohloff [18] completed the structural determination by establishing the stereochemistry of the irone isomers they had isolated from the Italian iris oil (probably from Iris pallida) first used by Ruzicka. They found that the oil contained the following four isomers (Fig. 6): (+)-cis-α-irone ((+)-4a), (+)-trans-α-irone ((+)-4b), (+)-β-irone ((+)-5), and (+)-cis-γ-irone ((+)-6a). Later on, in the same oil Rautenstrauch and Ohloff [19] were able to detect traces of the trans-γ-irone together with some other isomers, probably showing Z stereochemistry at the double bond in the side chain.

It has been established that the distribution of irone isomers and enantiomers in different qualities of iris oils depends upon the botanical species of the plant [10, 12, 19, 20]. The average composition [21] of an iris butter prepared from Iris germanica is the following: 0.91% of (+)-trans-α-irone (ee = 96%, (+)-4b), 61.48% of (–)-cis-α-irone (ee = 82%, (–)-4a), 0.71% of β-irone (5) and 37.60% of (–)-cis-γ-irone (ee = 38%, (–)-6a). When Iris pallida rhizomes are used, the corresponding iris butter contains 4.16% of (+)-trans-α-irone (ee = 98%, (+)-4b), 34.46% of (+)-cis-α-irone (ee = 66%, (+)-4a), 0.16% of β-irone (5) and 61.26% of (+)-cis-γ-irone (ee = 96%, (+)-6a).
Commercial racemic irones do not satisfy under the organoleptic point of view the demand of iris fragrance of modern perfumery. The natural extract is still used in perfume compositions. Several enantioselective syntheses have been reported in the literature [22–29] for the enantiomerically enriched irone isomers, but none of them has found practical application. During the last four years we were able to prepare all the ten isomers of irone in enantiopure form by enzyme-mediated approach, starting from the commercial mixture of racemic cis and trans-α-irone (Irone alpha®, Givaudan) [30–33]. The synthetic schemes leading to the preparation of all these enantiopure isomers are reviewed in this work.
2 Preliminary investigations
In the aim of using the same approach we had successfully employed to prepare enantiomerically pure (R)- and (S)-α-ionone (7) [34], we submitted Irone alpha® to sodium borohydride reduction in methylene chloride/methanol solution, and obtained the four racemic α-irols 8–11 [31] (Fig. 7).

The reduction mixture was submitted to bio-catalysed acetylation in t-butyl methyl ether solution, in the presence of vinyl acetate as an acyl donor, using two different kinds of enzymic preparations: Porcine Pancreatic Lipase (Sigma, Type II), and Lipase PS Pseudomonas cepacia (Amano Pharmaceuticals Co.) (Table 1). When PPL was used as a catalyst, a 2:1 mixture of cis- and trans-α-irol acetates was obtained (24-h reaction time). The main component (60.2%) was enantiopure derivative ent-15, showing a rather good diastereoisomeric enrichment (de = 78%.). The unreacted alcohol still contained 4.3% of stereoisomer ent-11 (chiral GC of the corresponding acetates).
Stereoisomeric composition of the enzyme-produced α-irol acetates. a Chirasil DEX CB column.
| Enzyme | Acetylated product (area%, chiral GC a ) | Unreacted alcohol (area%, chiral GC a ) |
| PPL | ent- 12 (25.4), (+)- 13 (7.6), (+)- 14 (6.8), ent- 15 (60.2) | 8 (16.0), ent- 8 (11.7), (–)- 9 (16.1), (+)- 9 (14.0), (–)- 10 (12.9), (+)- 10 + 11 (25.0), ent- 11 (4.3) |
| Cis/trans ratio = 2/1 | ||
| ent -12 ee = 99%, de = 54% | ||
| ent -15 ee = 99%, de = 78% | ||
| Lipase PS | ent -12 (27.1), (+)- 13 (27.1), (+)- 14 (22.4), ent -15 (22.4) | 8 (26.9), (–)- 9 (27.1), (–)- 10 (22.5), 11 (22.5) |
| Cis/trans ratio = 1.2/1 | ||
| ent -9 ee = 99%, de = 0 | ||
| ent -12 ee = 99%, de = 0 |
Lipase PS-mediated acetylation proceeded rapidly, to afford a nearly equimolar mixture of the four (9R) diastereoisomers ent-12, (+)-13, (+)-14, and ent-15 in enantiopure form. The enzyme neither discriminated between trans- and cis-α-irols, nor showed any diastereoselection within each single set, i.e. ent-12 vs (+)-13 or (+)-14b vs ent-15. The unreacted material recovered from the reaction was an equimolar mixture of the four enantiopure (9S) alcohol stereoisomers 8, (–)-9, (–)-10, and 11 (24-h reaction time). We use the prefix ent to distinguish the enantiomers of those derivatives for which the sign of the optical rotatory power could not be determined.
The results of these lipase-mediated reactions showed that separation of cis- and trans-α-irone derivatives had to be accomplished before the enzymic step.
3 Epoxydation of commercial Irone alpha® [30]
On the basis of a previous experience on α- and β-ionone epoxides [35], we found that key intermediates for the separation of trans- and cis-α-irone were the corresponding epoxides. Treatment of Irone alpha® with 3-chloroperbenzoic acid in methylene chloride gave an oily mixture, containing two main products (±)-16 and (±)-17 (Fig. 8), which could be separated by column chromatography. Derivative (±)-17 was obtained in pure form by crystallisation from hexane. X-ray single crystal analysis allowed us to establish that this crystalline material was the diastereoisomerically pure epoxide derivative of cis-α-irone bearing the oxirane ring in anti relation with the side chain at C(6).
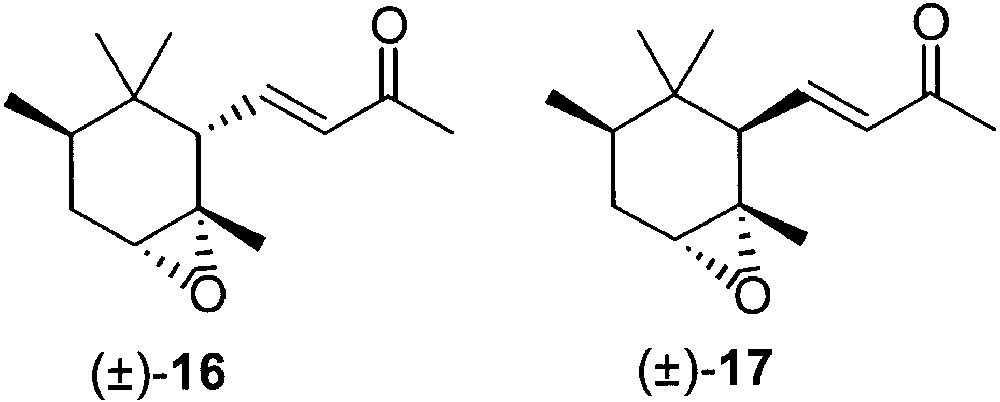
The first eluted fraction of the epoxidation mixture was composed mainly by (±)-epoxy trans-α-irone 16, with 5% of the other diastereoisomer of epoxy-cis-α-irone, i.e. the one obtained by reaction of the peracid on the same side of the substituent at C(6). As a matter of fact, the treatment of this chromatographic fraction with trimethylchlorosilane and sodium iodide in acetonitrile [36] afforded a 95:5 mixture of trans- and cis-α-irone. The stereochemistry of the epoxy moiety of derivative 16 was determined by 1H NMR analysis of a suitable derivative.
3.1 Trans-α-irones
Substrate (±)-16, impure of the cis derivative, was reduced with sodium borohydride in methylene chloride/methanol to provide, as major components, the two racemic diastereoisomeric alcohols (1:1 ratio) (±)-18 and (±)-19, which could be separated by column chromatography (Fig. 9). The first eluted alcohol (±)-18 was obtained in pure form by crystallisation of the corresponding 3,5-dinitrobenzoate ester 20 from ethanol. This material was assigned the ring relative stereochemistry depicted in structural formula 20 on the basis of NMR studies.
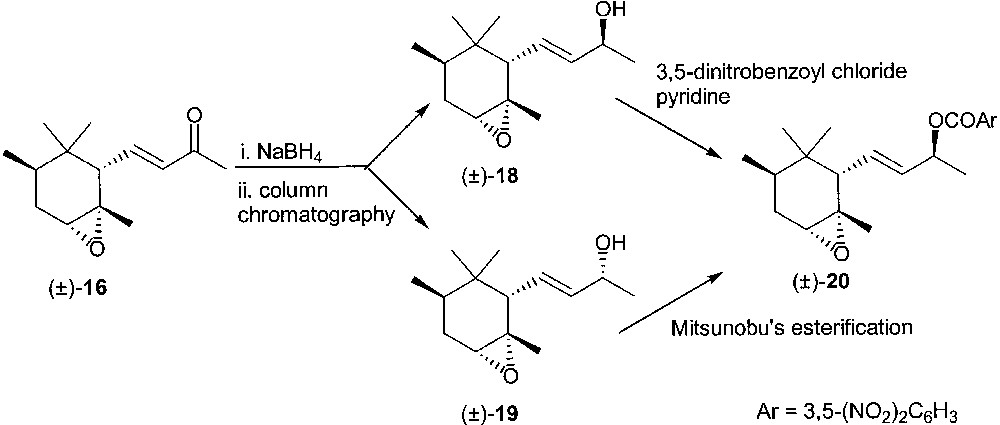
The 3,5-dinitrobenzoate ester of racemic 19 was an oil. Crude 19 was then esterified with 3,5-dinitrobenzoic acid under Mitsunobu conditions to provide 20. The two batches of crystalline ester 20 were collected, crystallised from ethanol and hydrolysed under basic conditions to afford (±)-18. Treatment of (±)-18 with vinyl acetate in t-butyl methyl ether in the presence of lipase PS Amano afforded enantiomerically pure acetate (+)-21 and unreacted alcohol (–)-18 showing ee = 98% (GC of the corresponding acetate derivative) (Fig. 10). The acetate (+)-21 was hydrolysed under basic conditions and the derived alcohol was converted into the epoxy ketone (+)-16 by reaction with MnO2 in methylene chloride solution. Similarly, alcohol (–)-18 afforded enantiopure (GC) (–)-16. The enantiomeric epoxy derivatives (–)-16 and (+)-16 provided upon deoxygenation (–)-trans-α-irone 4b (chemical purity = 88%, GC; ee = 98%, GC; [α]D20 = –400, c 1.05, CH2Cl2, lit. ref. [18] [α]D20 = –420, c 0.98, CH2Cl2) and (+)-trans-α-irone 4b (chemical purity = 96%, GC; ee = 98%, GC; [α]D20 = 427, c 0.95, CH2Cl2, lit. ref. [23] [α]D20 = 432, c 2.85, CH2Cl2, 86% ee) in good yields.
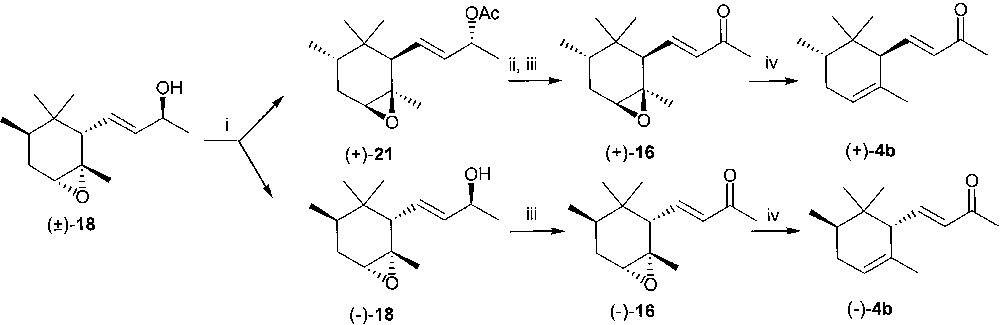
(i) Lipase PS, t-butylmethyl ether, vinyl acetate; (ii) KOH in MeOH; (iii) MnO2 in CH2Cl2; (iv) NaI, (CH3)3SiCl, CH3CN.
3.2 Cis-α-irones
More direct was the access to the enantiomers of cis-α-irone. Epoxy-cis-α-irone (±)-17 was reduced with sodium borohydride, to afford a 1:1 mixture of the two diastereoisomeric alcohols (±)-22 and (±)-23, which could be separated by column chromatography. The second eluted alcohol (±)-23 was then purified by crystallisation from hexane. In separate experiments (±)-22 and (±)-23 were submitted to Lipase PS mediated acetylation in t-butylmethyl ether solution in the presence of vinyl acetate, to provide, after column chromatography, acetates (+)-24 and (+)-25, and unreacted alcohols (+)-22 (ee = 98%, GC of the corresponding acetate derivative) and (–)-23 (ee = 98%, GC of the corresponding acetate derivative), respectively (Fig. 11). Acetate (+)-24 gave upon hydrolysis and oxidation with manganese(IV)oxide the same enantiomer of epoxy-irone 17 obtained by direct oxidation of (–)-23. The two samples of (–)-17 were gathered (ee = 98%, GC) and submitted to deoxygenation with trimethylchlorosilane and sodium iodide in acetonitrile, to give (+)-cis-α-irone 4a (chemical purity = 81%; ee = 98%, GC, [α]D20 = 117, c 1.5, CH2Cl2, lit. ref [18] [α]D20 = 111, c 0.92, CH2Cl2 ). Derivative (+)-17 (ee = 98%, GC), prepared both from acetate (+)-25 and from alcohol (+)-22, was transformed according to the same deoxygenation procedure into (–)-cis-α-irone 4a (chemical purity = 85%; ee = 98%, GC; [α]D20 = –130, c 1.55, CH2Cl2; lit. ref. [23] [α]D20 = –103.7, c 0.65, CHCl3, 86% ee).
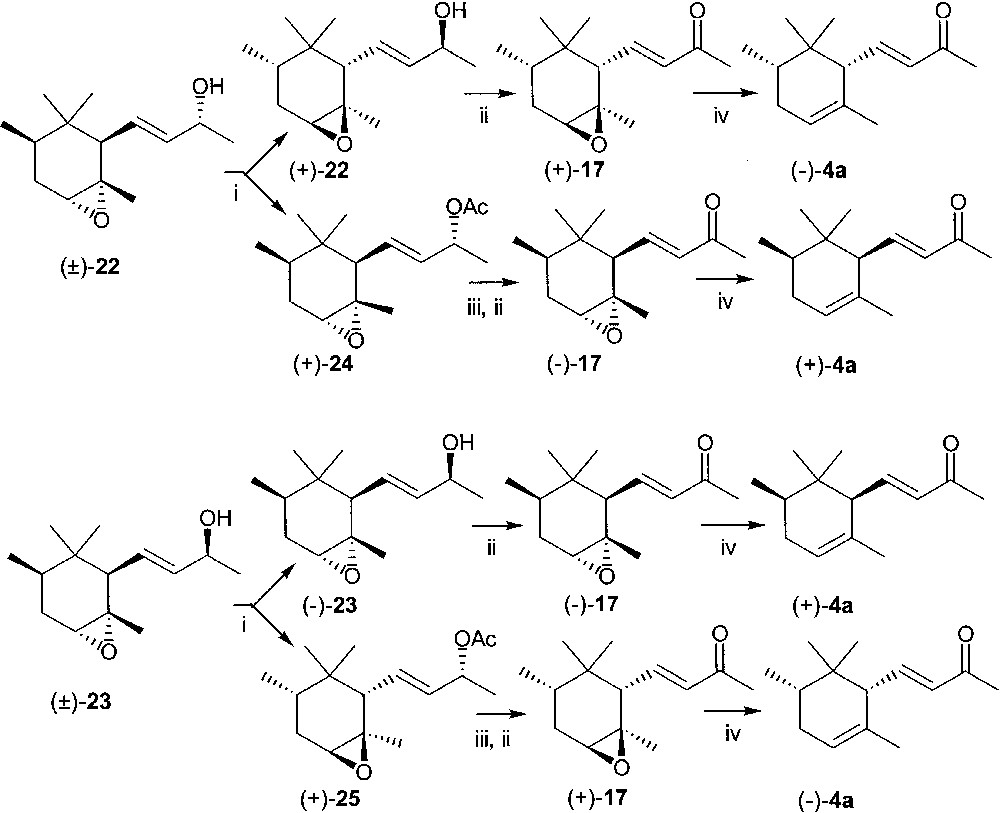
Lipase PS Amano, vinyl acetate, t-butylmethyl ether, column chromatography; (ii) MnO2 in methylene chloride; (iii) KOH in methanol; (iv) Me3SICl, NaI in acetonitrile.
4 Reduction of epoxy trans-α-irone and epoxy-cis-α-irone [32]
In order to develop a convenient synthetic approach to the other irone isomers, we thought useful to investigate the reductive opening of the 4,5-oxirane moiety of compounds 16 and 17 and the following dehydration.
4.1 β-irones
Treatment of racemic 16 with LiAlH4 in refluxing THF afforded diols (±)-26 and (±)-27 in ca. 1:1 ratio. These products crystallised from hexane, once separated by column chromatography. In both compounds, hydride attack at the less substituted carbon atom afforded the tertiary alcohol with the hydroxyl group on the same side of the side chain, through the highly favoured trans-diaxial opening [37] of the oxirane ring (Fig. 12).
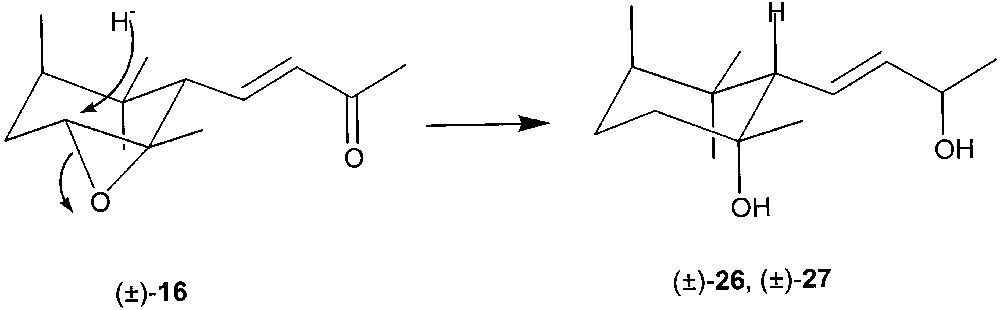
In enzymic-acetylation experiments, diols 26 and 27 were converted into the corresponding (9R) acetate esters (+)-28 and (+)-29, respectively, upon treatment with vinyl acetate in the presence of lipase PS in t-butyl methyl ether solution (Fig. 13).
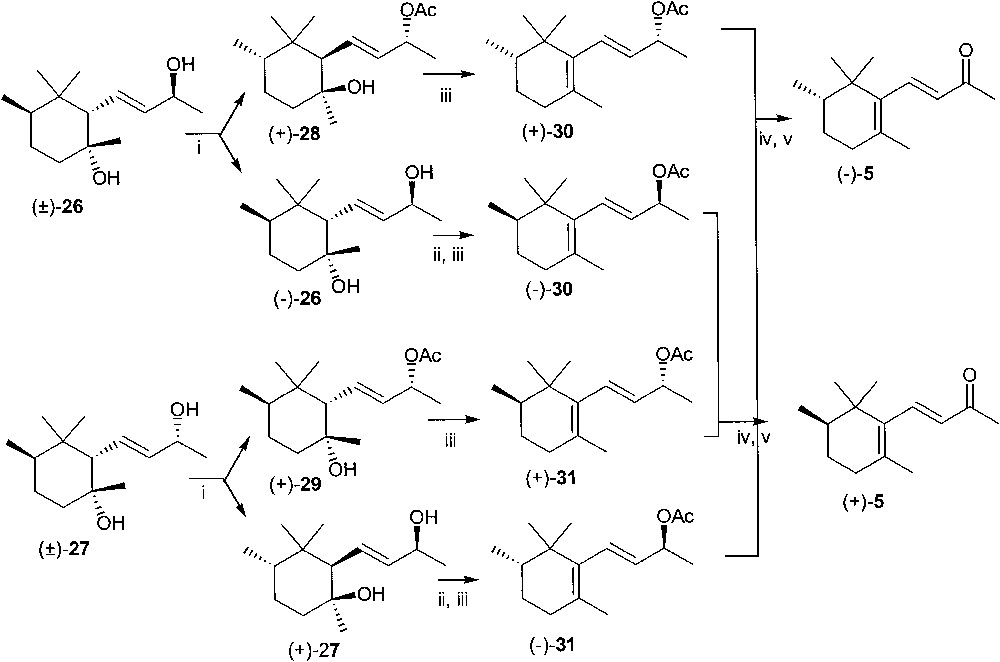
(i) Lipase PS, t-butylmethyl ether, vinyl acetate, column chromatography; (ii) acetic anhydride in pyridine; (iii) phosphorous oxychloride in pyridine; (iv) KOH in methanol; (v) manganese(IV) in methylene chloride.
The enantiomeric excesses of acetyl derivatives (+)-28 and (+)-29 and of the survived alcohols (–)-26 and (+)-27 were inferred from those of their transformation products, i.e. β-irol acetates. (+)-30, (+)-31, (–)-30 and (–)-31, respectively.
We first investigated the dehydration of tertiary alcohol moiety of allylic monoacetates 28 and 29 by reaction with POCl3 in pyridine, as it was reported to proceed with high stereo- and regioselectivity [38–42]. At room temperature, monoacetate (+)-28 reacted slowly with POCl3, the conversion being complete after one week. β-Irol acetate (+)-30 (ee = 99% chiral GC) was obtained in 90% yield, impure of 9% of (–)-trans-α-isomer. The γ-isomer was not detected by GC/MS amongst the reaction products. The components of the oily mixture could not be separated by chromatography and the same was true for the alcohols obtained by basic hydrolysis. A purification of β-irol from the contaminating trans-α-isomer was attempted through fractional crystallisation of the 4-nitrobenzoate ester, obtained as low melting crystals from cold hexane. However, the ratio of the isomers in the mixture recovered from the basic hydrolysis of the nitrobenzoate ester was substantially unaltered respect to that of the starting material.
The sequence described for (+)-28 was extended to (+)-29 and to the acetyl derivatives of (–)-26 and (+)-27, observing a similar distribution of isomers (Fig. 13). The (2R) diastereoisomeric β-irol acetate esters (–)-30 and (+)-31 were pooled, hydrolysed and oxidised with MnO2 to provide (+)-(2R) -β-irone ((+)-5) in high yield. Similarly, from (+)-30 and (–)-31 the (–)-(2S)-enantiomer ((–)-5) was obtained. (+)-5 and (–)-5 prepared through this synthetic sequence were contaminated by (–)-trans-α-irone (9%, GC–MS) and (+)-trans-α-irone (7%, GC–MS), respectively. This made the definition of the enantiomeric purity through optical measurements very difficult.
Reports on the characterisation of the enantiomers of β-irone were scarce. Mixtures highly enriched in β-irone could be obtained upon equilibration of the α- and γ-isomers [18, 43] in basic medium. Product (+)-5, containing a minute, but definite amount of (+)-cis-γ-irone 6, obtained via basic isomerisation of the natural extract originally used by Ruzicka for the structural elucidation studies, was assigned a specific optical rotation of +33 in CH2Cl2 (calculated value +59) as deduced by Rautenstrauch and Ohloff [18, 43]. Chapuis and Brauchli [23] found [α]D20 = –65.0° for the (–)-enantiomer 5 (86% ee), prepared by synthesis and purified by preparative gaschromatography. Unfortunately, our samples of (+)-5 and (–)-5 were contaminated by (–) and (+) trans-α-irone, whose specific optical rotations ([α]D20 = –400, c = 1.05 CH2Cl2 and [α]D20 = +427, c = 0.95 CH2Cl2 [30]) were opposite in sign and of much higher numerical value than those of the β-irone enantiomers. Taking into account the chemical composition of the two samples of β-irone [(+)-5: 91% β-irone and 9% (-)-trans-α-irone, GC–MS; (-)-5: 93% β-irone and 7% (+)-trans-α-irone, GC–MS], we determined an extrapolated value of specific optical rotation for enantiopure β-irone of ca. ±60 from the measured values of [α]D20 = +17.1 (c = 1.45, CH2Cl2) and [α]D20 = –20.2 (c = 2.30, CH2Cl2).
The enantiomeric purity of the two β-irone samples was then assessed by a spectroscopic NMR method based on the use of chiral shift reagents. By this method, the two samples of β-irone (+)- and (–)-5 were found to be enantiomerically pure
In order to verify whether a different regiochemical course of dehydration could be achieved, we experimented other reaction conditions, besides the concerted elimination of phosphate ester promoted by phosphorous oxychloride and affording β-irone. The tertiary alcohol (±)-29 did not react at room temperature by treatment either with pyridinium 4-toluenesulfonate in ethanol, or with 4-toluenesulfonic acid in toluene, or with thionyl chloride/pyridine in chloroform. However, it was soon converted into a complex mixture of decomposition products, by forcing the reaction conditions.
4.2 Cis-α and cis-γ-irones
(±)-Epoxy-cis-α-irone 17 showed a remarkable different behaviour under LiAlH4 reduction compared to racemic epoxy-trans-α-irone 16. The reduction of (±)-17 was much slower than of (±)-16, and afforded a ca 3:1 mixture of 4-hydroxy-4,5-dihydro-cis-α-irols 32 and 33 and 5-hydroxy-4,5-dihydro-cis-α-irols 34 and 35. Each component of the mixture was separated by column chromatography, the 4-hydroxy-isomers 32 and 33 being the first to be eluted.
This course of epoxide opening could be tentatively explained on the basis of a competition between the hydride attack at the most hindered carbon atom C(5), leading to a highly favoured trans diaxial opening in a chair-like transition state, and the reaction at the less substituted carbon atom C(4) (Fig. 14). The stereochemistry thus obtained and shown in the structural formulas of 32 and 33 was confirmed by 1H NMR analysis.

Derivatives 32–35 were submitted to enzymic acetylation with vinyl acetate and lipase PS, in t-butyl methyl ether solution, to provide (9R) acetate esters (+)-36, (+)-37, (+)-38 and (+)-39, respectively, and the (9S) allylic alcohols (+)-32, (–)-33, (+)-34 and (+)-35 (Figs. 15 and 16). Interestingly enough, the enzyme-mediated transesterification of 4,9-diols 32 and 33 afforded only the allylic (9R) acetate esters under these conditions.
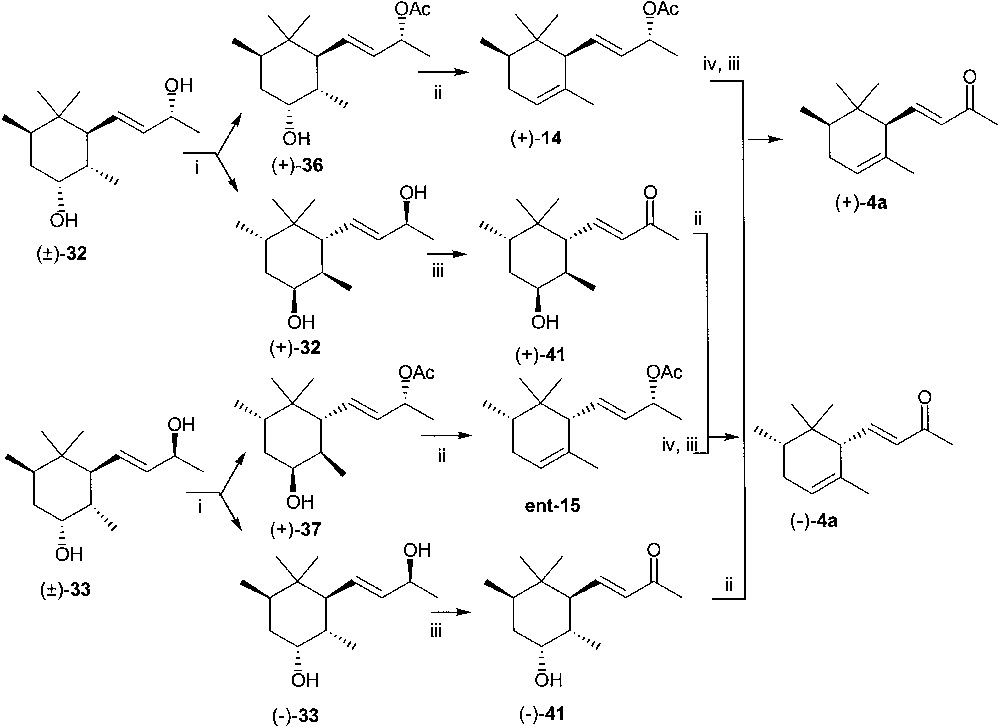
(i) Lipase PS, t-butylmethyl ether, vinyl acetate, column chromatography; (ii) phosphorous oxychloride in pyridine; (iii) manganese(IV) oxide in methylene chloride; (iv) KOH in methanol.

(i) Lipase PS, t-butylmethyl ether, vinyl acetate, column chromatography; (ii) acetic anhydride in pyridine; (iii) phosphorous oxychloride in pyridine; (iv) hν, xylene, isopropanol; (v) KOH in methanol; (vi) manganese(IV) oxide in methylene chloride.
The (9R) 4-hydroxy-4,5-dihydro-cis-α-irol acetate esters (+)-36 and (+)-37 and the corresponding (9S) alcohols (+)-32 and (–)-33 were converted into cis-α-irone 4a enantiomers through the convergent sequences outlined in Fig. 15. Treatment of the acetate esters (+)-36 and (+)-37 with POCl3 in pyridine at room temperature provided within a few minutes the diastereoisomeric α-irol acetate esters (–)-14 (ee = 98%, chiral GC) and ent-15 (ee = 97% chiral GC). These latter compounds were found to contain 13% (GC–MS) and 28% (GC–MS), respectively, of the corresponding 4-chloro derivatives, obtained by chlorine substitution of the intermediate phosphate ester (Fig. 17). (–)-14 and ent-15 afforded (+)-and (–)-cis-α-irone 4a by basic hydrolysis and MnO2 oxidation, making possible the purification of α-irone from the contaminating 4-chloro-4,5-dihydro-ketone analogue (+)-40 by column chromatography (Fig. 18). The conversion of diastereoisomeric 4,9-diols (+)-32 and (–)-33 into (–)- and (+)-4a by the same route required the regioselective chemical acetylation of the allylic alcohol. The attempt based on the use of acetic anhydride and pyridine in CH2Cl2 failed: ca. 1:1 mixtures of 4- and 9-mono acetate esters were invariably obtained. However, by inverting the order of application of the reactions of Fig. 15, (+)-32 and (–)-33 were straightforwardly converted into (–)- and (+)-4a. To this end, (+)-32 and (–)-33 were first oxidised with MnO2 to the enantiomeric 4-hydroxy-4,5-dihydro-cis-α-irones (+)-41 and (–)-41, then treated with POCl3 in pyridine.


This sequence would represent a new simple entry to enantiomerically pure forms of cis-α-irone from Irone alpha® via crystalline epoxy derivative 5. However, there is a drawback in the process. As a matter of fact, the POCl3/pyridine treatment of the 4-hydroxy derivatives (+)-36, (+)-37, (+)-41 and (–)-41 produced also the corresponding 4-chloro-4,5-dihydro analogues in small amount by nucleophilic substitution, close to the desired dehydration products with the cis-α-irone skeleton. The chlorinated compounds were separated by column chromatography and completely characterised as ketone derivatives (+)-40 (from (+)-36 and (–)-41) and (–)-40 (from (+)-37 and (+)-41). The stereochemistry depicted in the structural formulas of (+)- and (–)-40 was in accordance with the inversion of the configuration at C(4) promoted by the nucleophilic substitution of the phosphate ester with chloride anion (Fig. 17) and was confirmed by 1H NMR analysis.
The equatorially located chlorine atom could not be eliminated. Various reaction conditions were attempted: (CH3)3COK/THF, DBU/CHCl3 and 35% KOH/ethanol. Thus, the rapid dehydration of (+)-36, (+)-37, (+)-40 and (–)-41 occurring by POCl3/pyridine treatment was the result of the anti periplanar elimination of the axially located phosphate ester together with the axial hydrogen atom at C(5). The competing substitution of the phosphate ester with chloride anion occurred with inversion of configuration at C(4), and the desired 4,5-dehydrohalogenation became very difficult for stereochemical reasons.
The (9R) acetate esters (+)-38 and (+)-39 and the (9S) enantiomers, obtained upon treatment with acetic anhydride/pyridine of the alcohols (+)-34 and (+)-35, were also treated with POCl3/pyridine. These materials reacted immediately with POCl3 even at 0 °C, affording, almost quantitatively, mixtures of cis-γ-, cis-α- and β-irol acetate diastereoisomers in ca 45:35:20 ratio. Chiral GC analysis of the reaction mixtures showed conservation of the enantiomeric purity during the dehydration process.
The different regiochemistry of the POCl3/pyridine induced dehydration of the tertiary alcohols of the trans series (derived from (±)-26 and (±)-27) and of the cis series (derived from (±)-34 and (±)-35) could be tentatively explained, taking into account the precise stereochemical requisites of E2 elimination. Antiperiplanar arrangement of the hydrogen atom and of the other leaving group is required. In such cyclohexane derivatives, this steric condition is best satisfied when the hydrogen atom and, in this case, the phosphate ester are in trans diaxial relation. The analysis of the chair conformations reported in Figs. 12 and 14 allowed us to draw the following considerations:
- • in derivatives (±)-26 and (±)-27, the hydrogen linked to C(6) and the hydroxylic group at C(5) are in trans diaxial relation; thus, dehydration of the corresponding allylic acetates to β-derivatives is highly favoured; traces of trans-α-irol acetate are due to a competing antiperiplanar elimination with the axial hydrogen at C(4);
- • in derivatives (±)-34 and (±)-35; the alcohol function at C(5) is equatorial, and is eliminated to give cis-γ and cis-α derivatives preferentially. The distribution of the dehydration products might suggest a E1 mechanism [44].
The mixture of cis-γ-, cis-α- and β-irol acetate diastereoisomers, prepared from (+)- and (–)-38 and (+) and (–)-39, as well as the alcohols obtained by basic hydrolysis, resulted inseparable by SiO2 column chromatography. Thus, the amount of the γ-isomers present in the various mixtures was increased by photoisomerization, using conditions similar to those described in the synthesis of presiccanochromene A by Nozoe and Hirai [45] from an intermediate similar to our products. The irol acetate mixtures were thus irradiated with UV lamps, in an isopropanol solution containing 10% xylene, following the course of the reaction by GC/MS (Fig. 16). Within 24 h, the cis-α-irol acetate was completely converted into the corresponding cis-γ analogue, while the β isomer almost immediately disappeared, giving rise to various minor products which were not characterised. Before the conversion of the α-isomer could be complete, we noticed also the formation of the (7Z)-isomer of cis-γ-irol acetate. At the end of the photoisomerisation, SiO2-column chromatography allowed us to separate the mixture of (7Z)- and (7E)-cis-γ-irol acetates from the more polar products of transformation of the β-isomer. No further purification was possible by chromatography of the irol mixture obtained by basic hydrolysis of the latter acetate esters. During MnO2 oxidation, (7Z)-cis-γ-irone was formed together with the (E)-isomer, but it was the first to be eluted from the SiO2 column chromatography with 3–4% ethyl acetate in hexane, thus a final complete separation was possible.
All the mixtures originated from the POCl3/pyridine treatment of (+)- and (–)-38 and (+)- and (–)-39 were UV irradiated, to give 42 and 43 γ-irol acetate derivatives, pooled according to the stereochemistry at C(2) and C(6), hydrolysed in alcoholic potassium hydroxide, and then submitted to MnO2 oxidation and chromatographic purification (Fig. 16). The following samples of cis-γ-irone were obtained:
- • (+)-cis-γ-irone 6a (96% ee chiral GC), containing 5% (+)-cis-α-irone 4a and 3% of (+)-β-irone 5; [α]D20 = 9.5 c = 2.15, CH2Cl2;
- • (–)-cis-γ-irone 6a (97% ee chiral GC), containing 4.5% (–)-cis-α-irone 4a and 1.5% (–)-β-irone 5; [α]D20 = –6.3 c = 1.05, CH2Cl2.
Some discrepancies could be observed in the magnitude of the specific rotations of cis-γ-irone reported in the literature. The following data of [α] in solution were available: (i) [12]: (–)-cis-γ-irone, chemical purity (GC) 99%, [α]57820 = –1, c = 0.5, CH2Cl2; (+)-cis-γ-irone, chemical purity (GC) 99%, [α]57820 = +2, c = 0.5, CH2Cl2; (ii) [18] (+)-cis-γ-irone, [α]D20 = 2, c = 0.443, CH2Cl2; (iii) [23]: (–)-cis-γ-irone, purified from (–)-trans-γ-irone by preparative GC, ee = 76%, [α]D20 = –5.3, c = 1.66, CH2Cl2; (iv) [46]: (-)-cis-γ-irone [α]D20 = -5.3 c = 0.75, CHCl3; (v) [28] (+)-cis-γ-irone, de = 99%, ee = 99%, [α]D20 = +0.4, c = 3, CHCl3.
It was only possible to conclude that optically pure cis-γ-irone was characterised by a very low specific rotation. Our values, however, were altered by the presence of traces of cis-α-irone and β-irone showing specific rotations of the same sign. Enantiomeric purity was then determined by chiral GC analysis.
5 Photoisomerisation of α-irone derivatives [33]
5.1 Trans-γ and cis-γ-irones
We further exploited α-γ photoisomerisation to convert enantiopure trans- and cis-α-irol acetates into trans- and cis γ isomers. Key intermediates were the enantiomerically pure epoxy derivatives (–)-18 and (+)-21, (+)-22 and (+)-24, obtained by Lipase PS–mediated acetylation of (±)-18 and (±)-22, respectively, in t-butyl methyl ether solution in the presence of vinyl acetate as an acyl donor. Alcohol derivatives (+)- and (–)-18 and (+)- and (–)-22 were deoxygenated by reaction with zinc and sodium iodide in acetic acid at room temperature, to give (+)-9 and (–)-9 and (–)-10 and (+)-10. Acetylation of these derivatives in pyridine and acetic anhydride afforded α-irol acetates (–)-13 and (+)-13 and (–)-14 and (+)-14, in the trans and cis series, respectively. All the four α-irol acetates were submitted separately to photoisomerisation till the ratio between α- and γ-isomers, evaluated by GC–MS, did not change any more.
Isomerisation of trans-α-acetates (–)-13 and (+)-13 (0.04 M solution in isopropanol with 10% of xylene, Fig. 19) took ca 12 days: a photostationary equilibrium was reached, characterised by 2–7% of still unreacted α-isomer. The trans-γ-isomers obtained were found to be 1:1 mixtures of derivatives (–)- and (+)-44 and of the corresponding 7Z diastereoisomers 45 and ent-45 only when their 1H NMR spectrum was recorded. As a matter of fact, no distinction was possible by GC–MS.
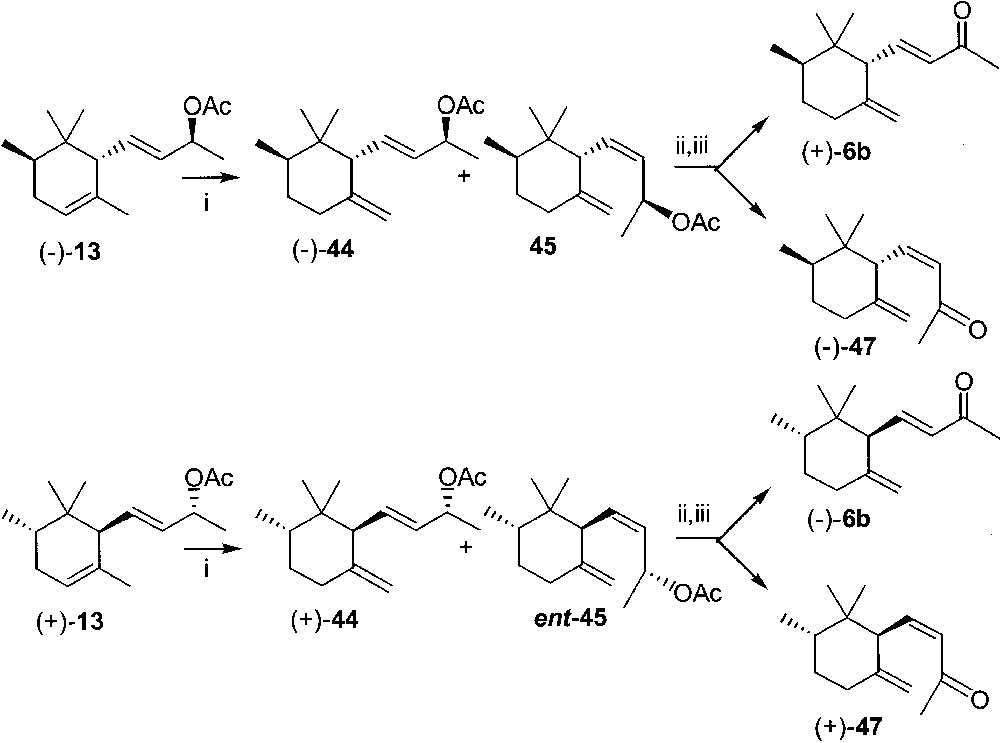
(i) hν, isopropanol, xylene; (ii) KOH, MeOH; (iii) manganese(IV) oxide, CH2Cl2, column chromatography.
As for a cis-α derivatives (+)-14 and (–)-14 (0.04 M solution in isopropanol with 10% of xylene, Fig. 20), after 5 h ca 40% of cis-γ-isomer (ent-42 and 42 respectively) was detected by GC, and no trace of 7Z isomers, either of the α or of the γ series, was found. Within 27 h from the beginning, cis-α-irol acetates had completely disappeared, and ca 30% of 7Z isomers 46 and ent-46b had formed. The appearance of (7Z)-isomers (5%) was first detected after an irradiation time of 7 h.

(i) hν, isopropanol, xylene; (ii) KOH, MeOH; (iii) manganese(IV) oxide, CH2Cl2, column chromatography.
The rate constants of the photoisomerisation processes were estimated from the disappearance (5%) of the corresponding α-irol acetate, as measured by GC/MS, using ca 5% of n-dodecane as an internal standard: ktrans = 2 × 10–6 s–1; kcis = 6 × 10–5 s–1. Owing to inherent design limitations of the reactor, constant temperature could not be maintained. A difference of ca. one order of magnitude was observed between the rate constants of cis and trans-α-irol acetate isomerisation. This could be tentatively attributed to the so-called ‘steric factor’. This process is probably very sensitive to the molecular geometry of the substrate itself, since triplet–triplet transfer generally needs a collision between sensitizer and substrate.
The photoisomerisation mixtures were hydrolysed in methanolic potassium hydroxide, and the corresponding irol derivatives were oxidised in methylene chloride solution in the presence of manganese (IV) oxide. Both in the trans and in the cis series, 7E-irone could be separated from 7Z diastereoisomer by column chromatography on SiO2. The following products were thus obtained:
- • from (–)-13: (7E)-(+)-trans-γ-irone ((+)-6b): [α]D20 = 10, c = 2.8 CH2Cl2, chemical purity = 74% (GC–MS), with 8% of (–)-trans-α-irone; ee = 99% (HPLC); (7Z)-(–)- trans-γ-irone ((–)-47): [α]D20 = –43.2, c = 1.25, CH2Cl2 (lit. ref. [25] [α]D20 = –42, c = 0.4, CH2Cl2, ee 76%); chemical purity = 70% (GC–MS);
- • from (+)-13: (7E)-(–)-trans-γ-irone ((–)-6b): [α]D20 = –16, c 3.0 CH2Cl2, chemical purity = 79% (GC–MS), with 3% of trans-α-irone; ee = 99% (HPLC); (7Z)-(+)-trans-γ-irone ((+)-47): [α]D20 = 48.7, c = 1.32 CH2Cl2; chemical purity = 76% (GC–MS).
- • from (–)-14: (7E)-(–)-cis-γ-irone ((–)-6a): [α]D20 = –2.54, c = 4.9 CH2Cl2; chemical purity = 96% (GC–MS), ee = 99% (GC); (7Z)-(+)-cis-γ-irone ((+)-48): [α]D20 = 50.5, c = 1.08 CH2Cl2, chemical purity = 90% (GC–MS).
- • from (+)-14: (7E)-(+)-cis-γ-irone ((+)-6a): [α]D20 = 1.78, c = 4.3 CH2Cl2, chemical purity = 93% (GC–MS), ee = 99% (GC); (7Z)-(–)-cis-γ-irone ((–)-48): [α]D20 = –48.3, c = 1.2 CH2Cl2, chemical purity = 91% (GC–MS).
This approach allowed us to obtain all the eight stereoisomers of E- and Z-cis- and trans-γ-irone from commercial Irone alpha. However, in the samples of (+)- and (–)-6b, the presence of α-isomers, showing high values of optical rotatory power of the opposite sign [(+)-4b [α]D20 = 427, c = 0.95 CH2Cl2; (–)-4b [α]D20 = -400, c = 1.05 CH2Cl2] could not be avoided, and strongly affected the values of the measured optical rotatory power. An alternative synthetic path affording enantiopure (+)- and (–)-6b with high chemical purity was thus devised.
This latter approach was based on the resolution of (±)-trans-γ-irone by enzymic methods. We thus optimised a large-scale synthesis of this racemic material, which is depicted in Fig. 21. Commercial 2,3-dimethyl-2-butene was brominated with NBS in CCl4 solution, in the presence of catalytic amounts of dibenzoyl peroxide, to give the bromo derivative 49 [47]. This latter was employed as alkylating agent of ethyl acetoacetate to afford 50, according to a general procedure for the alkylation of dianions of β-keto esters, reported in reference [48]. Cyclisation of substrate 50 with SnCl4 in methylene-chloride solution allowed us to obtain the known cyclic keto ester 51 [49], as a 2:1 mixture of trans/cis diastereoisomers, which had been previously used in a synthesis of racemic trans-γ-irone [50]. This derivative was submitted to reaction with PPh3 = CH2, followed by reduction of the ester function with lithium aluminium hydride, to afford trans-6-methyl-γ-cyclogeraniol (52) as a single diastereoisomer (GC–MS, 1H NMR). Oxidation to corresponding aldehyde and condensation with PPh3 = CHCOCH3 completed the synthetic path to racemic trans-γ-irone (only 2% of cis-γ isomer, GC–MS).
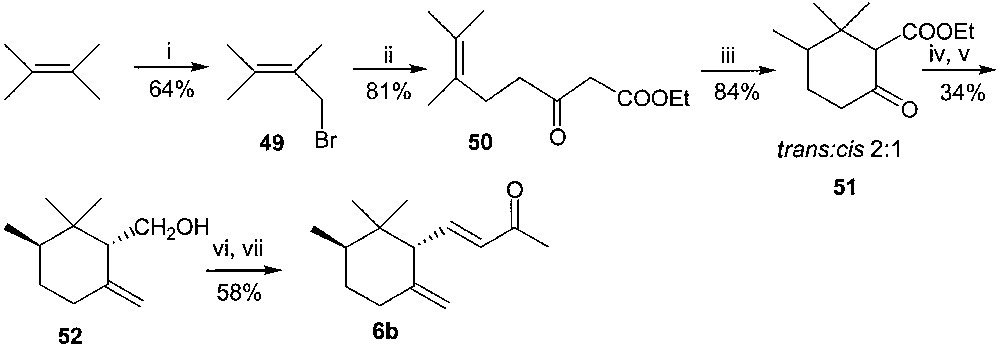
(i) NBS, CCl4, benzoyl peroxide; (ii) ethyl acetoacetate, 1 equiv NaH, THF, then 1 equiv BuLi, 0 °C; (iii) SnCl4, CH2Cl2; (iv) PPh3=CH2, THF, reflux; (v) LiAlH4, THF, 0 °C; (vi) ClCOCOCl, DMSO; Et3N, CH2Cl2; (vii) PPh3=CHCOCH3, toluene, reflux.
As for the resolution of the so obtained (±)-trans-γ-irone (Fig. 22), we took advantage of a scheme we had already exploited for α- [34] and γ-ionone [51]. Reduction with sodium boron hydride afforded a 1:1 mixture of γ-irols (±)-53a and (±)-53b, which were converted into the corresponding crystalline 4-nitrobenzoate esters (±)-54a and (±)-54b. These latter derivatives could be separated by fractional crystallisation from hexane. The less soluble diastereoisomer (±)-54b, obtained as a precipitate from the first crystallisation, was crystallised twice and reached de = 92% (GC–MS). The mother liquors of the first crystallisation, surprisingly enough, afforded diastereoisomer (±)-54a with de 98% (GC/MS) after four recrystallisations. This latter was the major product, and was then hydrolysed to give (±)-53a.
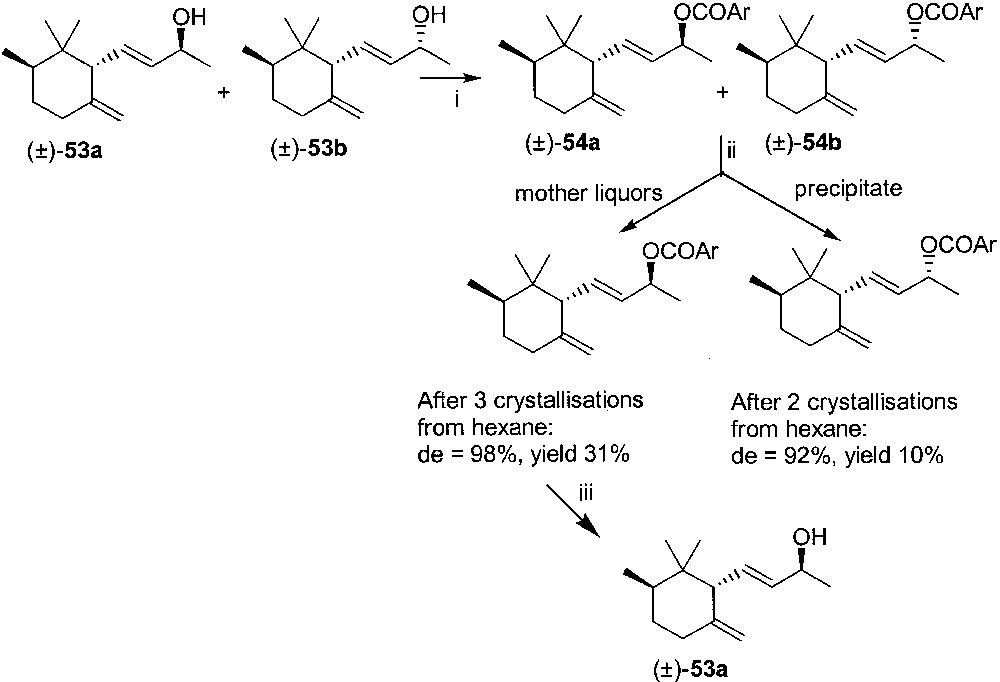
(i) 4-nitrobenzoyl chloride, pyridine; (ii) fractional crystallisation from hexane; (iii) KOH, MeOH.
After four days, Lipase-PS-mediated acetylation of racemic γ-irol 53a, in t-butyl methyl ether solution in the presence of vinyl acetate, gave γ-irol acetate (+)-44, and left unreacted alcohol (+)-53a (Fig. 23). Hydrolysis of (+)-44, followed by manganese (IV) oxide oxidation afforded (–)-trans-γ-irone: [α]D20 = –81, c = 1, CH2Cl2; chemical purity 99% (GC–MS); ee > 99% (chiral HPLC). Oxidation of the unreacted alcohol (+)-53a allowed to obtain (+)-trans-γ-irone: [α]D20 = 76.4, c = 1.2, CH2Cl2; chemical purity 99% (GC–MS); ee > 99% (chiral HPLC). The reference values of [α]D20 reported in the literature for (+)- and (–)-6b are rather low and not fully comparable: a) (–)-6b, ee 70% (NMR with chiral shift reagents), [α]D20 = –43.3 (c 0.8, CH2Cl2) [24]; b) (–)-6b, ee 76% (ee of the starting material), [α]D20 = –53.4 (c = 2.34, CHCl3) [23]; c) (+)-6b, ee 80% (ee of the starting material), [α]D20 = 57 (c 0.33, CH2Cl2) ref. [25]; d) (+)-6b, ee 99% (HPLC), [α]D20 = 59.4 (c 1.2, CHCl3) [27].
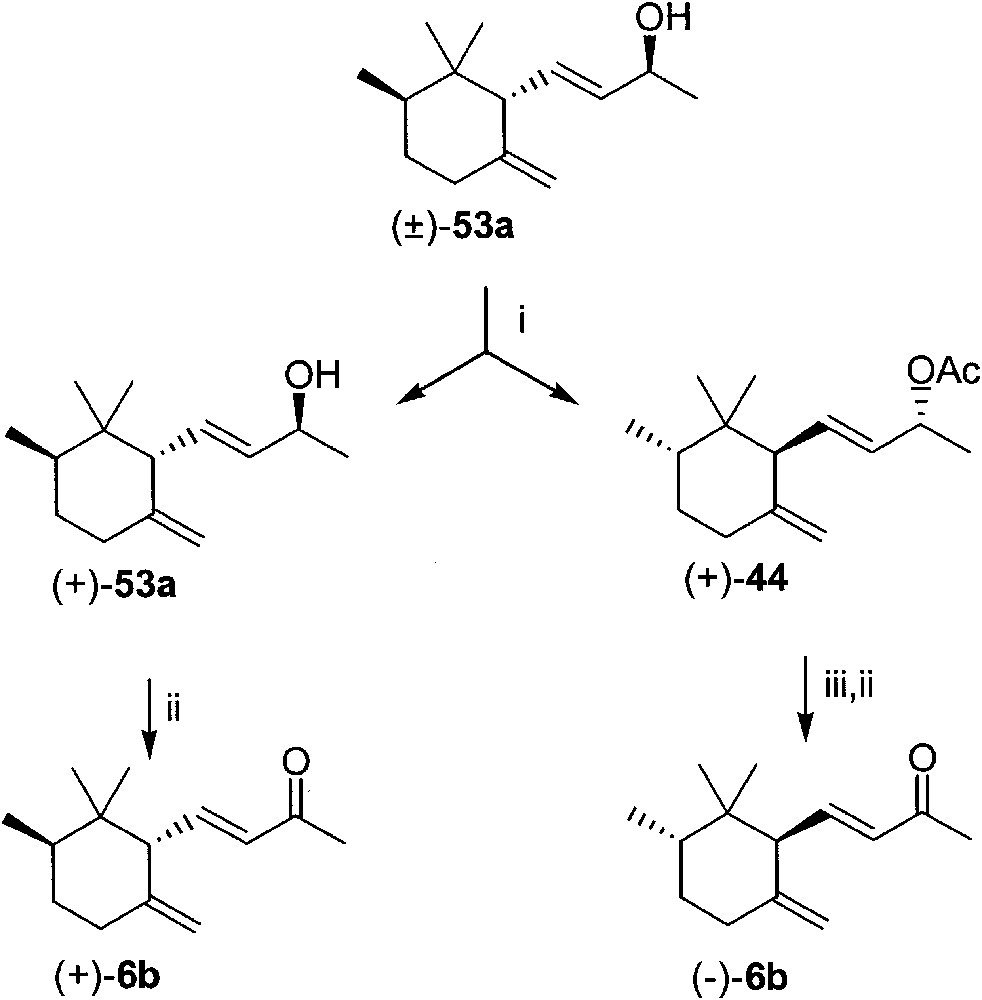
(i) Lipase PS, t-butylmethyl ether, vinyl acetate, column chromatography; (ii) MnO2 in methylene chloride; (iii) KOH in methanol.
6 Olfactory evaluations
All the enantiopure samples of irone isomers were submitted to olfactory evaluation and the following results were obtained:
- • (+)-4a, violet-like, with woody, methylionone undertones; odour threshold (od. thr.). = 100 ppm by triangular text;
- • (–)-4a, slightly stronger with a distinct ‘orris-butter’ character; od. thr. = 10 ppm by triangular text;
- • (+)-4b, the weakest of the α-isomers;
- • (–)-4b, showing a weak violet/wood/red berry character; neither (+)- nor (–)-trans-α-irone possesses the characteristic ‘orris’ odour;
- • (+)-6a, showing a floral, fatty, sweet, and woody odour character, an ionone-type odour with slightly sweet aspects; od. thr. > 100 ng l–1 by GC olfactometry;
- • (–)-6a, showing a β-ionone-type odour of warm floral-woody tonality; green aspects are present, too. It shows some fruity nuances, reminiscent of pineapples; the odour is linear, and it can be considered a dry-down note; od. thr. 0.75 ng l–1 by GC olfactometry;
- • (+)-6b, very weak, of a woody odour tonality; od. thr. 113.5 ng l–1 by GC olfactometry;
- • (–)-6b, not very powerful, but possessing a soft ‘orris-butter’- type of odour; od. thr. 26.35 ng l–1 by GC olfactometry;
- • (+)-5, possessing a β-ionone-type odour of warm floral-woody tonality with green and anisic aspects; the odour is linear, and the tenacity of the note is good.
- • (–)-5, having a woody odour with a distinct honey note, that is quite sweet; besides, it shows floral ionone-type facets, and a fruity tonality, but also an unpleasant smoky character; it belongs to the β-ionone-type family, without being very close to β-ionone.
These odour properties, evaluated on isolated samples, resulted to be quite different from those reported by Petrzilka et al. [21] and obtained by chiral GC sniff analyses of the corresponding racemic irones. According to this study, with the exception of trans-α-irone, all the other pairs of enantiomers could be distinguished by human nose. In particular, of cis-α and cis-γ-irone, only the (+)-enantiomers appeared to be odour active, showing also the finest and strongest iris-like notes.
The odour evaluations performed on our samples pointed out that all the irone isomers had different odour properties, and both (–)-cis-α- and (–)-cis-γ-irones were found stronger than the corresponding enantiomers. (–)-Cis-α-irone was also described to possess a distinct orris-butter character.
7 Conclusions
The discovery of irones dates back to more than one century ago: it was in 1893 when Tiemann isolated a ketonic odoriferous fraction from iris root oil. Since then, chemists have devoted plenty of work to establish their chemical constitution, configuration, and distribution according to the various iris species. A blind spot remained in their characterisation: the complete evaluation of the odour properties of all the ten irone isomers in enantiopure form as single isolated compound. This paper is a description of the efforts we devoted to reach this aim.
The great development had by enzyme chemistry during the last century and the commercial availability of enzyme preparations allowed us to successfully face the synthetic problem of irone isomers. We employed commercial racemic Irone alpha® as a starting material. We always obtained optical activation by lipase mediated kinetic resolution. By means of conventional organic reaction, i.e. epoxidation, reduction, dehydration, and photoisomerisation, we had access to α, β and γ irone skeletons. Thus, we had in our hands samples of enantiopure irones to submit to olfactory evaluation. The results of this study were found quite different from those obtained by Petrzilska by GC-sniffing. We found that (–)-cis-α-irone and (–)-trans-γ-irone showed the delicate orris-butter character. The odour response of all irone isomers was found to be extremely dependent on the relative and absolute configuration at the two stereocentres at C2 and C6.
Acknowledgements
Cofin-Murst is acknowledged for financial support. The authors are grateful to Robertet S.A. (Grasse, France) and Givaudan Dübendorf AG (Dübendorf, Switzerland) for the olfactory evaluations of irone samples.