

1 Introduction
The finding of efficient catalysts for the selective oxidation of organic molecules, under mild conditions, is a difficult challenge in organic chemistry. A possible strategy is to build biomimetic or bioinspired catalysts based on our detailed knowledge of the enzymatic systems that have been selected by living organisms for selective oxidations during life evolution. In that context, a huge amount of studies have been performed during these last 30 years on the use of chemical models to understand and to mimic a particular class of metalloenzymes, the cytochrome P450-dependent monooxygenases. These ubiquitous enzymes have attracted the interest of chemists for two main reasons: (i) they catalyze the selective monooxygenation by O2 of almost any organic substrate, including alkanes (according to Eq. (1)), and (ii) they play a key role in the oxidative metabolism of drugs and other xenobiotics, which is a crucial step in the adaptation of living organisms to their always changing chemical environment.
| RH + O2 + 2e− + 2H+ → ROH + H2O | (1) |
The huge number of studies that have been performed during the last three decades both on cytochromes P450 themselves and on iron porphyrin models has led to a detailed understanding of the oxidations catalyzed not only by P450s and bioinspired systems, but also by hemeproteins in general. It has also led to very important starting points for the understanding of the mechanisms of non-heme iron-containing oxygenases and for the design of bioinspired oxidation catalysts.
The main objective of this article is to give an overview of what were the main contributions of iron porphyrin model studies to the fields of cytochrome P450 and oxidation catalysis. This will be done through a brief comparative history of the development of our knowledge on cytochrome P450 chemistry and on iron porphyrin models, between 1975 and 2006.
The main contributions of chemical models to the field of cytochrome P450 and oxidation catalysis first concerned the understanding of (i) the structure and reactivity of the iron complexes involved in the P450 catalytic cycle, and (ii) the nature of the iron–metabolite complexes that are formed upon reaction of some substrates with cytochrome P450. They are also concerned with the development of metalloporphyrin-based systems that catalyze typical P450 reactions such as the hydroxylation of alkanes or aromatic compounds, the epoxidation of alkenes, N-, S- and O-oxidative dealkylations, N-oxidations, and sulfoxidations. Such systems have been successfully applied to the preparation of oxidized metabolites of drugs. Iron porphyrin-based systems have also been used to reproduce P450-catalyzed reactions different from monooxygenations, such as reductions or dehydrations. Sections 4–6 describe these different contributions of iron porphyrin models to the field of cytochrome P450 and oxidation catalysis, in an historical perspective. Before that, Section 2 gives an historical, brief overview of the development of our knowledge on cytochrome P450 itself, in order to better appreciate the impact of iron porphyrin model studies in that field. Section 3 shows how the 1975–1980 period has been crucial in the starting of the use of heme models to understand P450 reactions and to design biomimetic oxidation catalysts.
Cytochrome P450 and chemical model systems have been the subject of an intense research by a great number of laboratories in the world for these last 30 years. Obviously, this article will not provide a detailed review of all the involved topics. It will focus on the use of iron porphyrin models, even though Mn or Ru porphyrins have led to very interesting results in biomimetic oxidation catalysis. It will not analyze the structure and reactivity of porphyrin iron–oxo complexes and the mechanisms of substrate oxidations by these species, which have been the subject of several recent review articles. In a more general manner, for most of the treated subjects, it will mention recent review articles and will not give an exhaustive report of the data published on each subject.
2 Brief history of the progress of our knowledge of P450 chemistry in the 1960–2006 period
The existence of a CO binding pigment, that is reducible by NADPH and whose complex with CO exhibited an absorption maximum at 450 nm, was discovered in liver microsomes of pigs and rats by Garfinkel [1] and Klingenberg [2]. Six years later, this pigment was identified as a b-type cytochrome, containing iron protoporphyrin IX as cofactor (Fig. 1), by Omura and Sato [3]. The atypical position of the Soret peak of its CO complex at 450 nm was at the origin of the name given by the authors to this hemeprotein: cytochrome P450. During the following years, it was shown that this hemeprotein catalyzed the hydroxylation of various xenobiotics including drugs as well as steroid hormones, according to Eq. (1). Electrons that are necessary for these reactions were found to come from NADPH or NADH via electron transfer proteins, such as cytochrome P450 reductase, which are coupled to cytochrome P450. Many spectral and enzymatic studies, that were performed in the 1960s on liver microsomes or only partially purified preparations of P450, led to the proposition that P450 iron is bound to a protein cysteinate acting as an axial ligand. The first complete purification of a P450 was published in 1968; it concerned a bacterial P450 from Pseudomonas putida which hydroxylates camphor to 5-exo-hydroxycamphor [4]. Many studies performed on liver microsomal fractions and on this highly purified P450cam led, at the beginning of the 1970s, to a P450 catalytic cycle which was not far from the one admitted presently. These developments were reviewed in several articles and books, such as a book giving the proceedings of a symposium on microsomes and drug oxidations edited by Gillette et al. [5], and a book entitled Cytochrome P450 that was edited in 1984 by Rückpaul and Rein [6]. In order to have a detailed and complete view of the progress made on cytochrome P450 during these last 30 years, one should read the three editions of the book Cytochrome P450: Structure, Mechanism and Biochemistry, edited by P. Ortiz de Montellano that have appeared in 1986, 1995 and 2005 [7–9]. The first edition gives the state-of-the-art in the field in the mid-1980s, whereas the second and third editions show the fantastic development of this field during the last two decades.
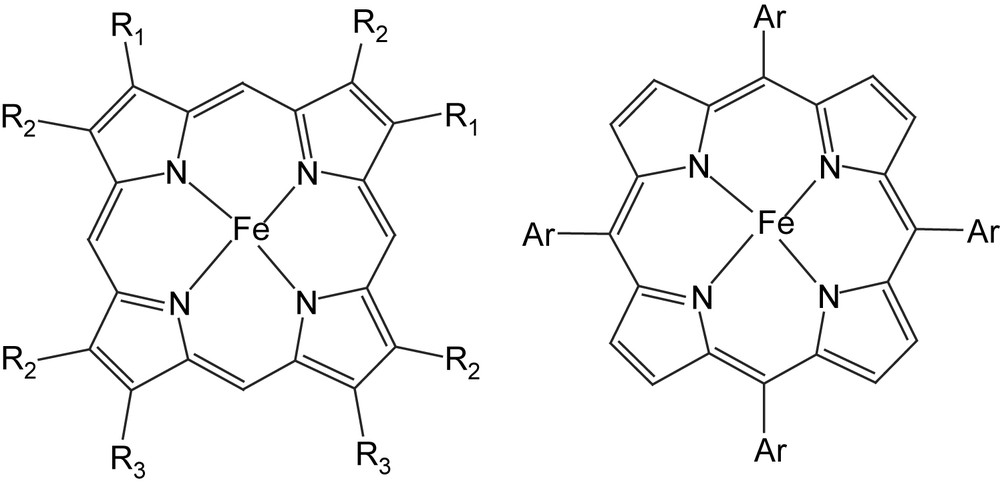
Formula of the main iron porphyrins mentioned in Sections 1–4. On the left, iron porphyrins derived from the heme: iron protoporphyrin IX, Fe(PPIX), R1 = CHCH2, R2 = CH3, R3 = CH2CH2COOH; iron protoporphyrin IX dimethylester, Fe(PPIXDME), R3 = CH2CH2COOCH3; iron deuteroporphyrin dimethylester, Fe(DPDME), R1 = H, R2 = CH3, R3 = CH2CH2COOCH3; iron beta-octaethylporphyrin, Fe (OEP), R1 = R2 = R3 = Et; on the right, synthetic iron meso-tetraarylporphyrins. Fe(TPP), Ar = C6H5; Fe(TMP), Ar = mesityl(2,4,6-trimethylphenyl); Fe(TDCPP), Ar = 2,6-dichlorophenyl; Fe(TF5PP), Ar = C6F5.
The intense research performed on P450s between 1965 and 2006 progressively revealed the huge number of P450s that exist in almost all living organisms such as mammals, fish, microorganisms and plants. Only a few protein primary sequences were available in 1985, whereas hundreds were published in 1995, and thousands are now available. Genome analyses recently showed that 57 genes are coding for P450s in humans, and that 246 genes are coding for P450s in the plant Arabidopsis thaliana [9]. Data accumulated on P450s also revealed the existence of several great diversities.
- • Diversity of the P450 biological functions, as P450s catalyze many reactions involved in the biosynthesis and biodegradation of important endogenous compounds, such as steroid hormones, fatty acid-derived mediators, or vitamins in mammals, and glucosinolates, isoflavonoids, and alkaloids in plants. They also play a key role in the metabolism of xenobiotics including drugs, and, consequently, in the adaptation of living organisms to their chemical environment.
- • Diversity of the reactions that they catalyze, as they not only catalyze monooxygenation reactions, which remain their most often encountered activities, but also several reactions without oxygen atom transfer from O2 to substrates, such as reductions, isomerizations, and dehydrations [10–12].
- • Diversity of the substrates that they can transform (see for instance Ref. [9] for an illustration of the extreme diversity of substrates admitted by human, plant and microbial P450s).
The first X-ray structure of a P450 was published in 1985–1987 [13]. It concerned the bacterial, soluble P450cam from P. putida and remained the paradigm for P450 structure–function studies until the structure of another soluble, bacterial P450 from Bacillus megaterium was solved in 1993 [14] (Fig. 2). Since then, many structures of P450s from various origins have appeared [15]. However, the first X-ray structure of a membrane-bound, mammalian P450, CYP 2C5 from rabbit liver, was only published in 2000 [16]. The first X-ray structures of mammalian P450–substrate complexes were reported three years later; they showed a great conformational adaptation of the active site of CYP 2C5 to different substrates such as diclofenac and a sulfaphenazole derivative [17,18]. Even more recently, between 2004 and 2005, the structures of four of the main human liver P450s were published [19].
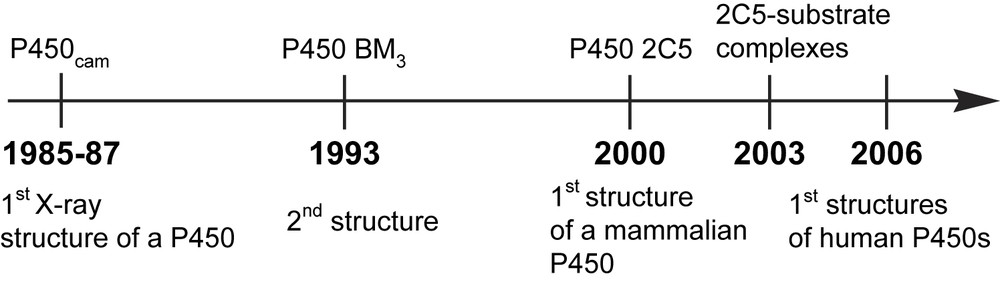
Some important dates in the development of our knowledge of P450 structures.
As mentioned above, the main steps of the P450 catalytic cycle of O2 activation and substrate hydroxylation were proposed in the early 1970s, on the basis of spectral and enzymatic studies performed on microsomal fractions and purified P450cam. They were confirmed and understood in more detail from many studies performed on various highly purified P450s. The use of a variety of biochemical and biophysical tools, such as mutagenesis, high-resolution cryocrystallography, and spectroscopic studies of intermediate states isolated using cryogenic or fast kinetic techniques, have led to the direct observation of P450 intermediates. They also led to a detailed understanding of the structural features of the protein that control the reduction and protonation of heme and dioxygen [20]. An important recent advance in that context has come from EPR and ENDOR cryospectroscopic studies of the P450 Fe(II)O2 complex after injection of an electron via γ-irradiation followed by thermal annealing of the sample [21]. This technique gave a first direct evidence for the successive formation of a Fe(III)–OO− species and a Fe(III)–OOH species. Fig. 3 shows the detailed P450 catalytic cycle of dioxygen activation that is derived from all these data (for more details see Ref. [20]).
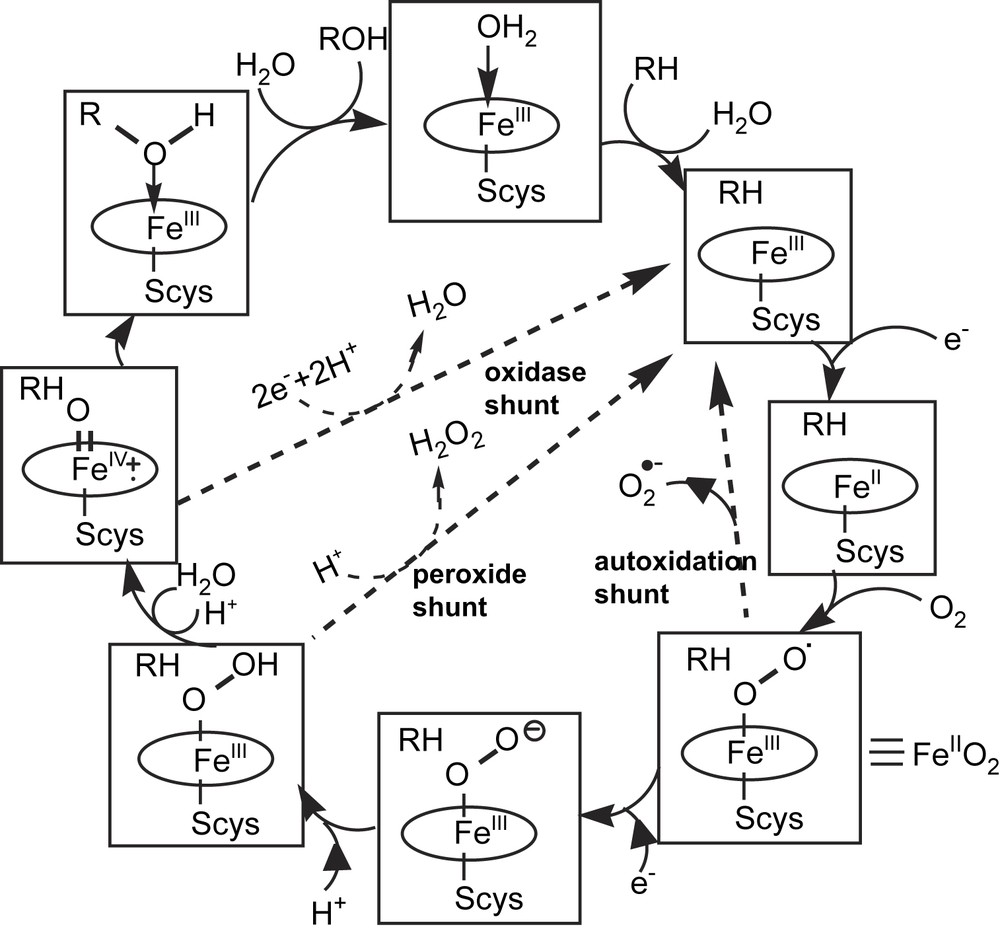
The P450 catalytic cycle of dioxygen activation and substrate hydroxylation (from Ref. [20]) RH = substrate; Scys = cysteinate ligand. Decoupling pathways resulting from oxidase activities of P450, in competition with the monooxygenase activity, are called autoxidation, and peroxide and oxidase shunts (- - ->).
The FeIII–OO− intermediate of this catalytic cycle has a nucleophilic character and appears to be the active species in some P450-catalyzed oxidative carbon–carbon bond cleavages of aldehydes that occur after the nucleophilic attack of the Fe–OO− species on the aldehyde function [10–12, 20]. There has been much discussion about the possible involvement of the FeIII–OOH intermediate in P450-catalyzed monooxygenations of some substrates such as olefins, amines or thioethers [22]. However, the hydroxylation of chemically inert C–H bonds requires a more reactive species, which is the P450 high-valent iron–oxo intermediate, and there is little doubt that this intermediate is mainly responsible for most P450-catalyzed monooxygenations.
Since the proposition of an oxygen rebound mechanism for P450-catalyzed C–H bond hydroxylations by Groves and McClusky [23], a great amount of experimental studies and calculations has been published on the mechanisms of these reactions (for most recent reviews see Refs. [20,22,24,25]). A linear, homolytic transition state, leading to the formation of a caged substrate-derived radical best fits the available data (Fig. 4). The use of ‘radical-clock’ substrates allowed one to measure the lifetime of the suspected radical cage intermediates. This lifetime of the radical intermediate critically depends on the tightness of the radical cage and all the steric and electronic constraints imposed by the active site on the radical within the cage. It should vary very much as a function of the nature of the substrate and of the P450 active site. Recent, detailed DFT calculations have suggested that the P450 iron–oxo active species, which should be a (porphyrin cation radical) Fe(IV)O complex comparable to peroxidase compound I [26], could have two quasi isoenergetic electron configurations, depending upon whether the unpaired electron of the porphyrin cation radical is ferromagnetically or antiferromagnetically coupled to the triplet Fe(IV)O species [24a]. These calculations are in favor of the linear transition state for C–H bond cleavage as shown in Fig. 4. However, they indicate different reaction coordinates for the fate of this transition state as a function of the electronic configuration of the iron–oxo species. With the high-spin state of this species, there is formation of a caged radical intermediate with the substrate-derived carbon radical interacting weakly with the Fe(IV)–OH species, in complete agreement with the mechanism shown in Fig. 4. The collapse of this intermediate to the product via formation of a C–O bond would require a significant energy barrier, whereas the reaction of the low-spin state of the FeO species could proceed to product without encountering such a barrier. This hypothesis of the two mechanisms dependent on two electronic configurations of the iron–oxo complex could explain the different mechanisms that were proposed on the basis of results observed with a variety of radical clock substrates (for a detailed discussion see Refs. [22,24]).
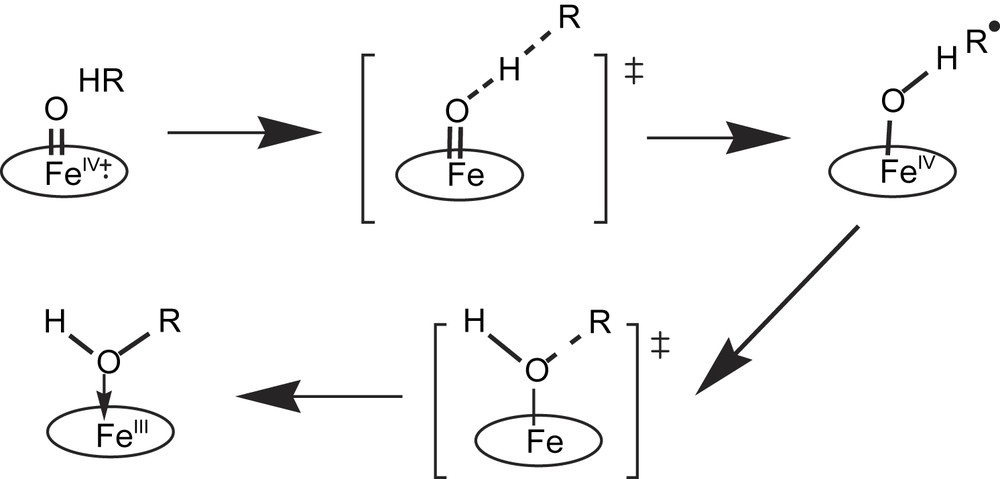
Most generally admitted mechanism for P450-catalyzed C–H bond hydroxylations (from Ref. [22]). This mechanism favors the formation of an intermediate caged radical derived from the substrate. Concerted insertion of the oxygen atom of the iron–oxo species into the substrate C–H bond, as well as intermediate formation of a substrate-derived cation R+ from electron transfer from R to the Fe(IV)–OH species, have also been proposed (see Refs. [22,24]).
3 The 1975–1980 period: starting of the use of iron porphyrin models to understand cytochrome P450 chemistry
It is during the 1975–1980 period that first data were published about the use of iron porphyrins (i) to mimic the intermediate stable iron complexes involved in the cytochrome P450 catalytic cycle, (ii) to understand the nature of several P450 iron–metabolite complexes, and (iii) to mimic the P450-dependent oxidation of substrates.
Thus, the first pentacoordinate Fe(III)porphyrin complexes involving an axial thiolate ligand, such as Fe(TPP)(SAr), Fe(OEP)(SR) and Fe(PPIXDME)(SAr), have been synthesized and completely characterized in the 1975–1976 period [27–30]. They exhibited UV–vis and EPR spectra very similar to the high-spin pentacoordinate ferric state of the P450 catalytic cycle (Fig. 3) (Table 1). Addition of various iron ligands to these pentacoordinate porphyrin Fe(III)–thiolate complexes, at low temperature, led to low-spin hexacoordinate Fe(III)(porphyrin)(SR)(L) complexes with L = alcohol, ether, imidazole, pyridine, n-octylamine, thioether, isocyanide, phosphine or thiolate. In all cases, there was a remarkable similarity between the spectral data of the low-spin Fe(III) model complexes and the corresponding cytochrome P450 Fe(III)–L complexes [31]. In that regard, very recent data on porphyrin Fe(III)(thiolate)(H2O) model complexes have led to a better understanding of the effects of the H2O ligand environment on the spin state of such models of the P450Fe (III) state [32]. In the 1975–1976 period, the synthesis of pentacoordinate Fe(II)(porphyrin)(alkylthiolate) complexes was also reported [33,34]. These complexes also exhibited spectral characteristics very similar to those of the high-spin pentacoordinate ferrous state of the P450 catalytic cycle (Table 1). As far as the hexacoordinate P450 Fe(II)–CO complex, whose absorption at 450 nm gave its name to P450, is concerned, Stern and Peisach first reported in 1974 the partial formation of a species characterized by a Soret peak at 450 nm when a thiol and a strong base were added to a ferrous heme in a DMSO–EtOH solvent in the presence of CO [35]. One year later, it was shown that quantitative formation of Fe(II)(porphyrin)(RS)(CO) complexes, that exhibited Soret peaks between 450 and 460 nm, occurred upon addition of alkylthiolate salts, as crown-ether complexes, to Fe(II)porphyrins under a CO atmosphere [36,37]. The great similarity of the spectroscopic characteristics of these iron porphyrin model complexes of the pentacoordinate Fe(III), hexacoordinate Fe(III), pentacoordinate Fe(II) and hexacoordinate Fe(II)CO states of the P450 catalytic cycle with those of the corresponding P450 complexes themselves (Table 1) was a key argument in favor of the presence of an axial cysteinate ligand in the P450 complexes. In that regard, one must keep in mind that the first X-ray structure of a cytochrome P450, which definitely confirmed the presence of a cysteinate axial ligand, was only published in 1986–1987 [13].
Comparison of spectral data of iron complexes of the P450 catalytic cycle and of some of their porphyrin models
| Complex | λ (nm) | EPR g-values | Year | Ref. | |||||
| P450cam Fe(III) (high-spin) | 391 | 520 | 540 | 646 | 8 | 4 | 1.8 | ||
| Fe(III)(PPIXDME)(pClC6H4S) | 396 | 533 | 571 | 643 | 7.2 | 4.8 | 1.9 | 1976 | [30] |
| P450cam Fe(III) (low-spin) | 417 | 535 | 571 | 2.45 | 2.26 | 1.91 | |||
| Fe(III)(PPIXDME)(pNO2C6H4S)(2Me–THF) | 418 | 535 | 566 | 2.36 | 2.26 | 1.94 | 1976 | [30] | |
| P450cam Fe(II) | 408 | 540 | |||||||
| Fe(II)(PPIXDME)(nBuS) | 408 | 540 | 560 | 1975 | [37] | ||||
| P450cam Fe(II)(CO) | 365 | 447 | 550 | ||||||
| Fe(II)(PPIXDME)(nBuS)(CO) | 376 | 458a | 552 | 1976 | [37] |
a Wave length of 449 nm in 50/50 DMSO–butanol instead of 458 nm in DMSO.
It is in the 1977–1979 period that iron porphyrins were used for the first time to understand the nature of P450-iron–metabolite complexes whose formation has been detected, between 1971 and 1974, by UV–vis spectroscopy during the metabolism of some substrates. Thus, studies on the reduction of polyhalogenated compounds related to CCl4 by iron porphyrins in the presence of a reducing agent in excess led to the first synthesis of porphyrin Fe(II)–carbene complexes [38–41] (Eq. (2)). These data, that were very important to interpret the nature of the P450 iron complexes, characterized by Soret peaks between 450 and 460 nm and formed during the reductive metabolism of polyhalogenated compounds, have been obtained only a few years after the spectral detection of those P450 complexes [42–45]. In a similar manner, the nature of the P450 iron–metabolite complexes, whose formation has been detected during the oxidative metabolism of several compounds involving an amine or N-hydroxylamine function [46], was interpreted on the basis of the synthesis and characterization of the first porphyrin Fe(II)–nitrosoalkane complexes that were published only a few years later [47] (Eq. (3)).
| (2) |

Finally, it is in the last part of the 1975–1980 period that the first successful biomimetic epoxidations of alkenes and hydroxylations of alkanes catalyzed by iron porphyrins were reported. Here too, a few years after the report that P450 catalyzed the monooxygenation of substrates by iodosylbenzene [48], it was shown that Fe(TPP)Cl catalyzed the epoxidation of cis-stilbene and the hydroxylation of cyclohexane and adamantane by PhIO [49,50] (Eq. (4)). In the same period, other potential oxygen atom donors, alkylhydroperoxides, were found to hydroxylate C–H bonds of various substrates in the presence of catalytic amounts of either P450 [51–54] or iron porphyrins [55,56]. It is noteworthy that first results, showing that O2 in the presence of a reducing agent (borohydride in that case) [57a], or ClO− [57b,c] is able to oxidize alkanes and alkenes in the presence of a catalytic amount of Mn porphyrins, were published in 1979–1980.
Actually, a few years before, systems associating an iron porphyrin with either a thiol and a base in large excess [58–61], or H2O2 [62] were found to oxidize substrates that are already chemically reactive by themselves such as anilines.
4 Use of iron porphyrins to determine the nature of stable P450 iron–metabolite complexes formed during the metabolism of some substrates
4.1 Complexes involving an iron–carbon bond
4.1.1 Iron–carbene complexes
4.1.1.1 Iron–carbene complexes formed upon reduction of polyhalogenated compounds
Historically, the first reported models of stable P450 iron–metabolite complexes were obtained during a study of the reduction of polyhalogenated compounds by Fe(II)porphyrins in the presence of a reducing agent in excess. Such a study had been undertaken to understand the nature of the P450–metabolite complexes formed upon reduction of CCl4 [42] and other polyhalogenated compounds such as CBr4, CCl3F, CCl3CN, CHCl3 and CHBr3 [45] by microsomal P450s in the presence of either dithionite or NADPH under low O2 pressures. These P450 complexes were characterized by Soret peaks between 450 and 465 nm (Eq. (6)).
The first porphyrin FeII–carbene complex, FeII(TPP)(CCl2), was obtained from the reaction of Fe(TPP)Cl with CCl4 in the presence of iron powder in excess under anaerobic conditions [38]. It was characterized by UV–vis, IR, 1H and 13C NMR spectroscopies, mass spectrometry, elemental analysis, and by an X-ray analysis of FeII(TPP)(CCl2)(H2O), 2DMF [39]. A very similar method, except for the use of sodium dithionite instead of iron powder as a reducing agent, allowed one to obtain a series of Fe(TPP) complexes with dihalogenocarbenes, CF2, CFCl, CFBr, CCl2 and CBr2 [63], and with halogenocarbenes bearing either an electron-withdrawing substituent (CClCN, CClCOOEt [40], CClCF3 [64]) or an electron-donating substituent (CClCH3, CClCH2OH, CClCHOHCH3, CClCHOHC6H5 [65]) (Eq. (5)). It was also possible to obtain a complex of Fe(TPP) with the monosubstituted carbene CHSC6H5 [66]. Table 2 summarizes all the porphyrin Fe(II)–carbene complexes that have been obtained by this method and completely characterized by various spectroscopic techniques, as well as the P450 complexes whose formation was detected by UV–vis spectroscopy upon reduction of the same polyhalogenated compounds. All these (TPP)FeII–carbene complexes are low-spin, diamagnetic species, sensitive to dioxygen and poorly reactive towards alkenes [63] except after photochemical activation [67].
| (5) |
| (6) |
(TPP)FeII–carbene complexes synthesizeda and P450 complexes spectrally detected, upon reduction of polyhalogenated compounds
| Starting compound | CRR′ carbene bound to (TPP)FeII | Refs. | P450 FeII–metabolite complex detected λmax (nm) | Refs. | |
| R | R′ | ||||
| CCl4 | Cl | Cl | [38,39] | 460 | [42,45] |
| CCl3F | Cl | F | [63] | 453 | [45] |
| CCl2F2 | F | F | [63] | –b | |
| CCl3CN | Cl | CN | [40] | 468 | [45] |
| CCl3COOEt | Cl | COOEt | [40] | – | |
| CBr4 | Br | Br | [63] | 465 | [45] |
| CBr3F | Br | F | [63] | ||
| CCl3CF3 | Cl | CF3 | [64] | ||
| CCl3CH3 | Cl | CH3 | [65] | – | |
| CCl3CH2OH | Cl | CH2OH | [65] | – | |
| CCl3CHOHCH3 | Cl | CHOHCH3 | [65] | – | |
| CCl3CHOHPh | Cl | CHOHPh | [65] | – | |
| CCl3SPh | Cl | SPh | [66] | – | |
| CCl3SCH2Ph | Cl | SCH2Ph | [66] | – | |
| CHCl2SPh | H | SPh | [66] | – |
a The indicated Fe(TPP)(carbene) complexes were completely characterized by various spectroscopic techniques.
b Means that the reduction of the corresponding polyhalogenated compound by P450 has not been studied.
The Fe(TPP)(CCl2) complex was the first-described dihalogenocarbene complex of a transition metal and, as mentioned above, complexes of Fe(TPP) with several dihalogenocarbenes, such as CF2, CFCl, CFBr and CBr2, could be obtained by the same method. The corresponding CI2 complex could not be prepared from reduction of CI4, as it further reacted with (TPP)FeII with the eventual formation of the μ-carbido complex [(TPP)Fe]2C [68] (Eq. (7)). When this reaction between two iron porphyrins was prevented by using an iron ‘basket-handle’ porphyrin, a stable Fe(basket-handle porphyrin)(CI2) complex could be easily isolated and characterized [63].
| (7) |
In a similar manner, the reduction of DDT, CCl3CHAr2 with Ar = pClC6H5, by Fe(TPP) in the presence of dithionite in excess did not stop at the stage of the expected carbene complex Fe(TPP)(CClCHAr2), because of a very easy elimination of HCl that led to the vinylidene carbene complex Fe(TPP)(CCAr2) [69] (Eq. (8)).
| (8) |
4.1.1.2 Iron–carbene complexes formed upon oxidation of benzodioxole derivatives
Benzodioxole derivatives, some of which are used as insecticide synergists [70], are known as strong inhibitors of P450-dependent monooxygenases, as they are oxidized by these enzymes to species that coordinate tightly to their heme iron [71,72]. The resulting ferrous P450 complexes exhibit a difference visible spectrum with a Soret peak at 455 nm which may be compared to the spectra obtained for P450 Fe(II)–CO and P450 Fe(II)–carbene complexes formed during reduction of polyhalogenated compounds. It had been proposed that the benzodioxole-derived P450 complexes could involve an iron–carbene bond formed during the oxidation of the benzodioxole CH2 group by the P450 iron–oxo intermediate ([73], see also Ref. [74] for a recent review on this subject, Eq. (9)).
The synthesis and characterization of corresponding model iron porphyrin complexes provided supporting evidence for this proposition of a carbene complex [41,75,76]. Thus, the Fe(TPP)(1,3-benzodioxol-2-carbene)complex was prepared by the reduction of 2,2-dichloro-1,3-benzodioxole by Fe(TPP) in the presence of a reducing agent in excess, as the carbene complexes described in the previous paragraph, and was completely characterized. Addition of n-butylthiolate to this complex at −10 °C led to the hexacoordinate Fe(II)(TPP)(carbene)(nBuS−) complex that exhibited a Soret peak at 459 nm, very close to that reported for P450 iron–benzodioxole-derived metabolite complexes [41] (Eq. (10)).
Interestingly, very recent data have shown that such a P450–carbene complex is formed during the metabolism of the drug paroxetine, explaining the clinical reports of potent cytochrome 2D6 inhibition by this drug [77].
4.1.2 Iron-σ-alkyl and aryl complexes
4.1.2.1 Formation from reduction of halogenated compounds
Further studies, performed in the 1980–1985 period, on the reductive metabolism of some halogenated compounds by P450s have shown the formation of P450 iron–metabolite complexes characterized by Soret peaks even more red-shifted than those of P450 Fe(II)–carbene complexes. This was found in the case of benzyl halides [78] (Eq. (11)) and of the anaesthetic agent halothane [43,79,80] (Eq. (13)) that led to P450 complexes absorbing between 470 and 480 nm. Iron porphyrin model studies have played a key role in our understanding of the nature of these complexes. They showed that the reduction of benzyl halides [81] (Eq. (12)) and of halothane [64] (Eq. (14)) by Fe(TPP) in the presence of dithionite led to low-spin ferric σ-alkyl complexes whose spectral characteristics were in good agreement with those of (TPP)Fe(III)-σ-alkyl complexes prepared by more conventional methods [82] (for other reviews on porphyrin iron-σ-alkyl complexes, see also Refs. [83–85]).
| (11) |
| (12) |
A detailed study of the 470 nm absorbing P450 complex derived from halothane showed that it was a low-spin ferric species (from EPR spectroscopy) and that its decomposition led to CHClCF2, in complete agreement with a σ-alkyl P450 Fe(III)–CHClCF3 structure [86] (Eq. (13)).
| (13) |
| (14) |
4.1.2.2 Formation from oxidation of hydrazines
At about the same period (1982–1983) the formation of similar P450 iron–metabolite complexes, exhibiting such unusually red-shifted Soret peaks around 480 nm, has been reported to occur upon the oxidative metabolism of alkyl- and aryl-hydrazine RNHNH2 [87,88] (Eq. (15)). Model reactions have shown that Fe(III)porphyrins such as Fe(TPP)Cl readily react with alkyl- and aryl-hydrazines, in the presence of a limited amount of an oxidant such as O2 or FeCl3, with formation of Fe(III)-σ-alkyl (or aryl)complexes (Eq. (16)) [89]. The σ-aryl complexes were much more stable towards O2 than the corresponding σ-alkyl complexes. This big difference of stability was also observed for the corresponding P450 complexes, explaining in part why the P450 Fe(III)–Ph complex derived from PhNHNH2 could be detected in rat liver several days after treatment with this hydrazine [90].
| (15) |
| (16) |
Almost simultaneously, the formation of ferric σ-aryl complexes was reported to occur upon in situ oxidation of aryl-hydrazines by several other hemeproteins, such as hemoglobin [91,92], myoglobin [92], and catalase [93]. The formation of σ-aryl complexes of prostaglandin synthase by the same method was published 11 years later [94]. The definitive proof for the existence of a hemeprotein Fe(III)–Ph bond was published in 1984, with the X-ray structure of the myoglobin Fe(III)–Ph complex [95] (Eq. (17)). The X-ray structure of the P450cam Fe(III)–Ph complex was published six years later [96].
| (17) |
Studies of the one-electron oxidation of the porphyrin Fe(III)–R complexes have revealed an interesting reaction that has important consequences in biology, the migration of the R axial ligand from the iron to a pyrrole nitrogen atom of the porphyrin ring. Actually, oxidation of porphyrin Fe(III)–R complexes either with a limited amount of oxidant such as FeCl3 or O2, or electrochemically, led to the corresponding Fe(II) N-substituted (R) porphyrin complexes [97–99]. In a similar manner, the one-electron oxidation of porphyrin Fe(II)–carbene complexes, that may be formally written as [Fe(II) ← CCR′] or [Fe(IV)CRR′], was shown to give an iron-N-substituted porphyrin with the carbene CRR′ moiety bridging the iron and a pyrrole nitrogen atom [100,101]. These data could be interpreted by the intermediate formation of unstable Fe(IV)–R or Fe(V)CRR′ species that rapidly undergo the reductive elimination of two cis iron ligands [102] (Eqs. (18) and (19)).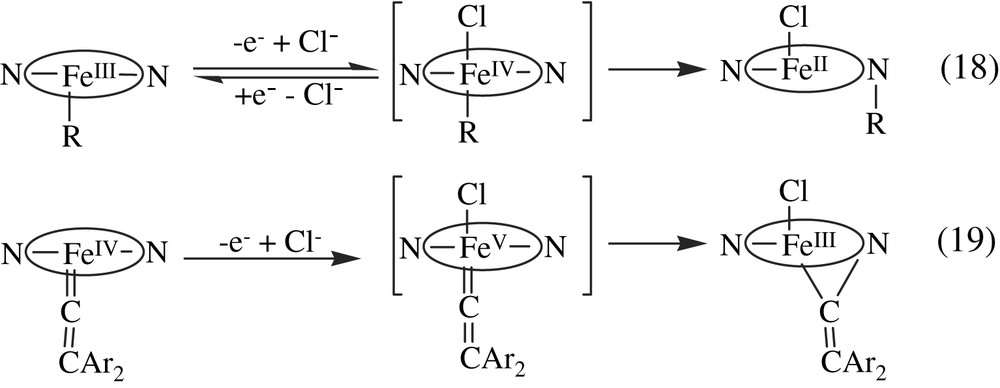
Such migration of σ-aryl (or alkyl) iron ligands to a pyrrole nitrogen atom also occurs in hemeproteins, as first-described in the case of hemoglobin [91]. This migration of σ-aryl P450 iron ligands was successfully used for topological analyses of the active site of a great number of cytochromes P450 (for a recent review on this subject see Ref. [74]). It has been invoked to explain the eventual formation of N-substituted hemes (green pigments) derived from P450s after oxidative metabolism of alkyl- and aryl-hydrazines and hydrazones, 1-aminoaryltriazoles, and sydnones [74].
4.1.3 Generality of the formation of iron–carbon bonds in P450 and iron porphyrin chemistry
The data shown in Sections 4.1.1 and 4.1.2 illustrate the very general formation of iron–carbon bonds during iron porphyrin- and P450-dependent oxidation or reduction of substrates. It seems that whenever a substrate-derived carbon-centered free radical is formed in close proximity of an iron(II)porphyrin, during such reactions, it may bind to the iron with formation of an iron(III)–R complex. This situation occurs after reduction of halogenated compounds by iron porphyrins in the presence of a reductant, or after oxidation of alkyl- and aryl-hydrazines by ferric porphyrins in the presence of an oxidant. The fate of such iron(III)–R complexes depends on the possible reactions of their iron–carbon bond in the conditions of their formation (Fig. 5).

Formation and possible fates of porphyrin iron-σ-alkyl or -aryl complexes.
Under anaerobic and aprotic conditions, the FeIII–R complexes are generally stable, whereas most of them are rapidly decomposed in the presence of protons or dioxygen with formation of the starting ferric porphyrin. In the presence of an oxidant, their one-electron oxidation may lead to a Fe(IV)–R complex which undergoes a migration of its R ligand to a pyrrole nitrogen atom as discussed above. Finally, in the case of polyhalogenated compounds (RX = X2CR1R2) and in the presence of a reductant, the intermediate [Fe(II)–R]− complex presumably formed from one-electron reduction of the Fe(III)–R complex should easily eliminate X− leading to the observed iron–carbene (Fe(II) ← CR1R2) complexes. These reactions are general; they have been observed with iron porphyrins and P450s as well as with other hemeproteins, such as myoglobin, hemoglobin or catalase at least for substrates that are small enough to come in close proximity of the heme.
The formation of iron–carbene complexes during metabolic oxidation of benzodioxole substrates by P450s suggests that such σ-alkyl Fe–R complexes could be formed as intermediates in P450-catalyzed oxidations [102–104]. A mechanism very often proposed for P450-catalyzed oxidation of alkanes and alkenes involves the intermediate formation of a substrate free radical derived from a hydrogen abstraction (C–H bond hydroxylation) or an addition (epoxidation) of the P450 high-valent iron–oxo species [22]. It is generally admitted that this intermediate free radical is then rapidly oxidized by the P450 Fe(IV) intermediate. This reaction could occur via a high-valent (formally Fe(V)) σ-alkyl P450 complex which would lead to P450 Fe(III) and the monooxygenated product by reductive elimination of the σ-alkyl and OH (or OR) ligands (Fig. 6). In the particular case of benzodioxole derivatives, there is another possible fate of the intermediate σ-alkyl complex, as 1,2-elimination of H2O could lead to the observed stable iron–carbene complexes [102–104]. In P450-catalyzed epoxidation of alkenes, a possible evolution of the high-valent iron-σ-alkyl intermediate could be the migration of the alkyl ligand to a pyrrole nitrogen atom, as one has seen above that porphyrin Fe(IV)–R complexes readily undergo such a migration of their R ligand (Fig. 5). Such reactions could be involved in the formation of green pigments (N-substituted hemes) observed during P450-catalyzed oxidation of terminal alkenes. Supplementary data are required to establish this mechanism ([102], in that context, see also Refs. [74,82–85]).
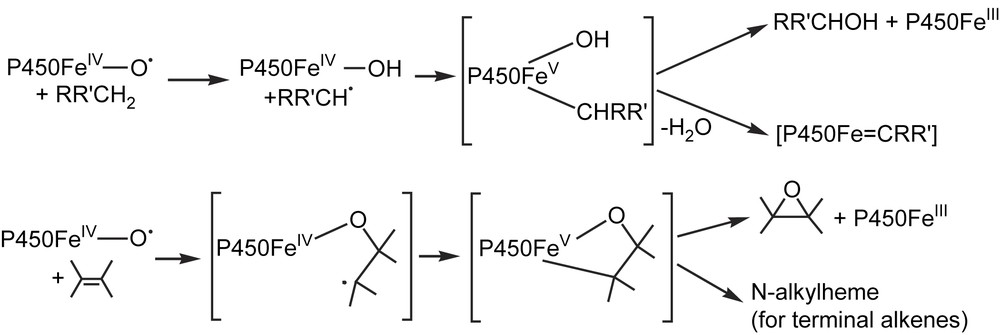
Possible intermediate formation of P450 iron-σ-alkyl complexes in C–H bond hydroxylation and alkene epoxidation.
4.2 Complexes involving an iron–nitrogen bond
4.2.1 Aza analogues of iron(II)–dioxygen complexes
In the 1970–1974 period, it has been reported that some amines, as well as an alkylhydroxylamine, N-hydroxyamphetamine, were oxidized to intermediate metabolites that coordinate tightly to P450 Fe(II) and gave rise to a spectrum characterized by a Soret peak around 455 nm [46]. As such complexes were formed after P450-dependent oxidation of alkylhydroxylamines RNHOH as well as after P450-dependent reduction of the corresponding nitroalkanes RNO2, it has been proposed that they were derived from the binding of nitrosoalkane intermediates to P450 Fe(II) [105–107] (Eq. (20)). Iron porphyrin model studies completely confirmed this proposition, as reaction of various N-alkylhydroxylamines RNHOH with Fe(III)porphyrins, and reaction of the corresponding nitroalkanes RNO2 with Fe(II)porphyrins in the presence of dithionite, led to very stable complexes involving a Fe(II)–NOR bond that were completely characterized by various spectroscopic techniques [47] (Eq. (21)).
The X-ray structure of one of them, Fe(TPP)(iPrNO)(iPrNH2), clearly showed that the nitrosoalkane ligand was bound to Fe(II) by its nitrogen atom [108]. Thus, these iron porphyrin model studies (i) allowed the first isolation of iron–nitrosoalkane complexes, (ii) strongly supported the existence of an iron(II)–nitrosoalkane bond in the amine-derived P450 iron–metabolite complexes, and (iii) underlined the great analogy between Fe(II)–O2 and Fe(II)–NOR bonds (RNO is isoelectronic to O2) [108] (Fig. 7). They also showed the great stability of porphyrin Fe(II)–NOR complexes, explaining why P450s engaged in such complexes are so strongly inhibited. Accordingly, it was later found that many drugs involving an amine function [109–111,74], such as antibiotics like troleandomycin and erythromycin [112–115] or the MAO inhibitor clorgyline [116], act as quasi-irreversible inhibitors of P450s, leading, in some cases, to severe problems of drug interactions [117].
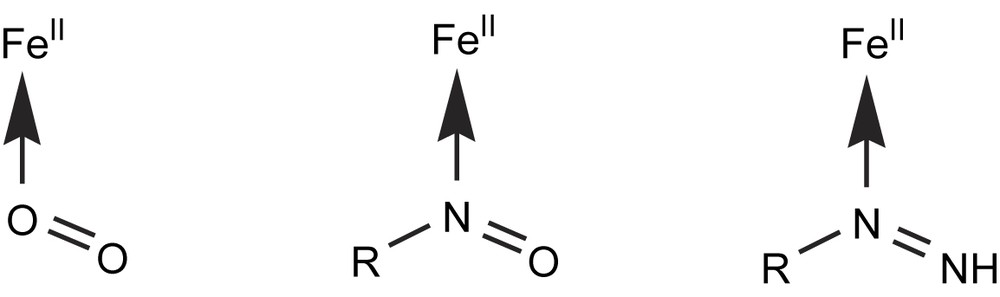
Analogy between iron(II)complexes of the isoelectronic O2, nitrosoalkanes and alkyldiazenes ligands.
The formation of Fe(II)–nitrosoalkane complexes is not limited to P450s and iron porphyrins. In 1977, it was reported to occur upon in situ oxidation of alkylhydroxylamines or reduction of nitroalkanes (in the presence of dithionite) by hemoglobin and myoglobin [118]. Later, it was reported to occur upon similar reaction with other hemeproteins such as prostaglandin synthase [119] and NO-synthase [120]. Even more recently, reaction of the NO-binding hemeprotein, guanylate cyclase, with CH3NO2 in the presence of dithionite was reported to lead to the nitrosomethane complex of this hemeprotein [121]. Recent review articles have been published on the interactions of organic nitrosocompounds with metals [122], and the crystal structure of the nitrosoethane complex of ferrous horse heart myoglobin was determined at a resolution of 1.7 Å [123]. This structure completely confirmed the proposition made on the basis of the structure of the Fe(TPP)(iPrNO)(iPrNH2) model complex [108] that nitrosoalkane ligands are bound to hemeprotein Fe(II) by their nitrogen atom. Some characteristics of the Fe(II)–N(O)R bond in the MbFe(II)–EtNO and iron porphyrin model complex are compared in Fig. 8.
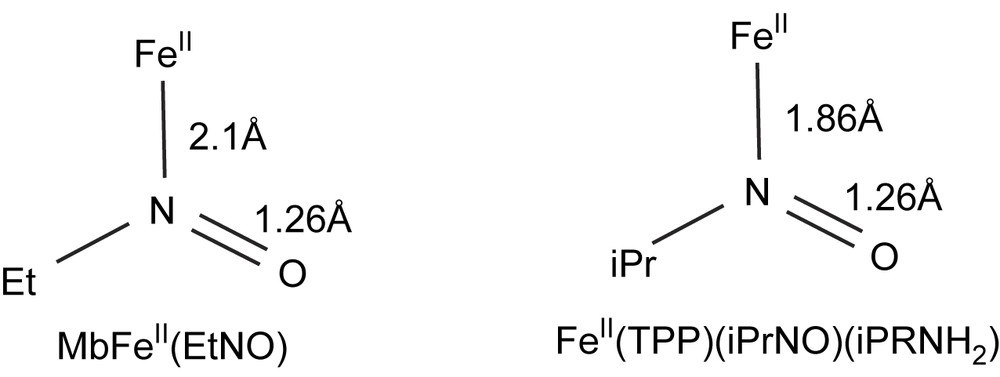
Comparison of some structural data of the myoglobin–iron(II)–nitrosoethane and Fe(TPP)(iPrNO)(iPrNH2) complexes (from Refs. [123] and [108], respectively).
Formation of another kind of aza analogues of porphyrin Fe(II)–dioxygen complexes, the Fe(II)–(NHNR) complexes (Fig. 7), has been detected by visible spectroscopy during the oxidation of alkylhydrazines by P450 [88], hemoglobin and myoglobin [124], and prostaglandin synthase [94]. Here too, the nature of these hemeprotein complexes was deduced from model studies of reactions between alkylhydrazines and iron porphyrins [89]. The presence of a Fe(II) ← NRNH bond in the isolated iron porphyrin complexes was deduced from their study by IR and 1H NMR spectroscopies [89]. Moreover, direct reactions of P450 Fe(II), myoglobin Fe(II), prostaglandin synthase Fe(II) and Fe(II) porphyrins with the corresponding alkyldiazenes led to identical Fe(II)–diazene complexes [88,124,94,89] (Eqs. (22) and (23)). However, so far, no X-ray structure determination was published for a porphyrin Fe(II)(RNNH) complex.
| (22) |
| (23) |
4.2.2 Aza analogues of high-valent iron–oxo complexes
P450 enzymes oxidize 1,1-disubstituted hydrazines, such as N-aminopiperidine to metabolites that coordinate tightly to the heme iron atom [125]. The corresponding complexes are characterized by Soret peaks around 450 nm in the ferrous state and around 438 nm in the ferric state. It has been speculated that these complexes are derived from the binding to P450 iron of the R2NN aminonitrene formed by in situ oxidation of the starting hydrazine R2NNH2 by the P450 active oxygen species [125] (Eq. (24)). The use of iron porphyrin models allowed one to understand these reactions, as the oxidation of 1,1-dialkylhydrazines by PhIO or O2 in the presence of iron meso-tetraarylporphyrins led to the preparation of (porphyrin) FeII–nitrene complexes [126] (Eq. (25)). These complexes were completely characterized by UV–vis, IR, 1H and 13C NMR, and Mössbauer spectroscopies [127].
The structure of the complex derived from oxidation of 1-amino-2,2,6,6-tetramethylpiperidine in the presence of iron meso-tetra-para-chlorophenylporphyrin has been fully established by an X-ray structure analysis [127]. It was also obtained by direct reaction of the iron(II)porphyrin with the free N-(2,2,6,6-tetramethylpiperidyl)-nitrene at −80 °C. It is a pentacoordinate iron complex with the aminonitrene as axial ligand. Its Fe–N–N sequence is linear, and its Fe–N(1.809 Å) and N–N(1.23 Å) bonds exhibit multiple-bond character. These nitrene complexes can be formally written as FeII ← NNR2 or FeIVNNR2 species. They are the aza analogues of the iron–oxo FeIVO intermediates formed in the catalytic cycles of several hemeproteins (Eq. (26)). However, it is noteworthy that 1H and 13C NMR and Mössbauer studies performed on the (porphyrin) iron–aminonitrene complexes are in favor of a well-defined high-spin ferrous (S = 2) state [127].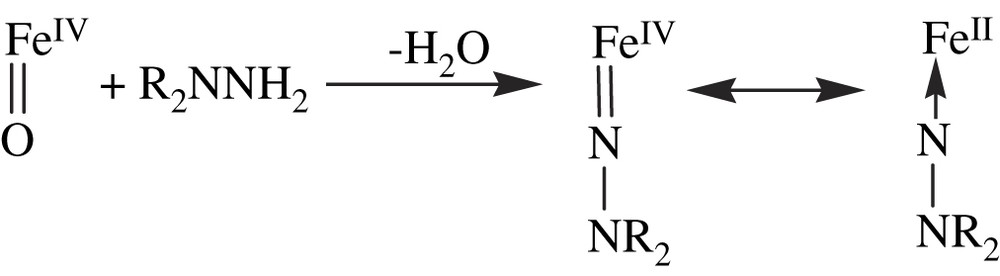
5 Catalytically active iron porphyrin model systems for cytochrome P450
5.1 Introductory remarks
When one considers the P450 catalytic cycle (Fig. 3), an ideal chemical model would associate an iron porphyrin, a thiolate ligand, a reducing agent and a source of protons, and O2 as oxygen atom donor. The use of a thiolate ligand appears to be difficult because of its too fast oxidation in such a strongly oxidizing medium. Anyway, during the development of oxidation catalysts bioinspired from P450, it rapidly appeared that the presence of an axial thiolate iron ligand was not required for efficient oxygenation of substrates. A very difficult problem in the design of chemical systems mimicking P450-catalyzed oxygenation of substrates by O2 in the presence of a reducing agent (according to Fig. 3) was to avoid a very fast competing reaction of the iron–oxo intermediate, the reduction of this species by the reducing agent usually present in large excess. It is why the best results reported so far were obtained for systems mimicking P450-catalyzed oxygenation of substrates by single-oxygen atom donors such as PhIO, according to a shortened P450 catalytic cycle only involving P450 Fe (III) and the P450 iron–oxo intermediate. As mentioned above (Section 3), the first successful model system for alkene epoxidation and alkane hydroxylation consisted of PhIO as oxygen atom donor and Fe(TPP)Cl as a very simple catalyst [49]. A major problem encountered with this first generation of metalloporphyrins as oxidation catalysts was their fast oxidative degradation in the medium. Major progress has been made in the development of efficient biomimetic oxidation catalysts thanks to an impressive number of studies that have been reported during the last 30 years. Many review articles and book chapters have been published on that subject [128–141]. For recent reviews one should see the book chapter of Groves et al. published in 2005 [22], and a review of Meunier et al. [142] that gives a detailed and complete view of the development of metalloporphyrins as oxidation catalysts between 1975 and 2000.
5.2 Important aspects of the development of efficient P450 biomimetic oxidation systems
5.2.1 Choice of the metal
A comparison of the porphyrin complexes of various transition metals led to select Fe, Mn, and Ru porphyrins as the best oxidation catalysts.
5.2.2 Choice of the best porphyrin ligands
The synthesis of three successive generations of porphyrin ligands eventually led to metalloporphyrin catalysts much more resistant towards oxidative degradation and more efficient for oxygenation of substrates. The early P450 models (1975–1980) used metal complexes of the easily accessible TPP (Fig. 1), as it was rapidly realized that meso-substituted metalloporphyrins were more stable than metal complexes of protoporphyrin IX, deuteroporphyrin IX, or β-octaethylporphyrin [50]. Introduction of bulky substituents on the ortho positions of each meso-phenyl ring, as in meso-tetramesitylporphyrin or in meso-tetra-(2,6-dichlorophenyl)porphyrin, TDCPP, led to a second generation of metalloporphyrins more resistant towards oxidative degradation because of an hindered approach of the oxidants to the very reactive meso-positions. Finally, the introduction of several electron-withdrawing substituents on the β-pyrrole positions of the second generation metalloporphyrins led to even more efficient catalysts, at least for some oxidations, because of an increase of the electrophilicity of their metal–oxo active species. Main representatives of this third generation of metalloporphyrins are shown in Fig. 9. Eq. (27) illustrates the improved efficiency of the third generation metalloporphyrins on the example of the hydroxylation of a poorly reactive alkane, heptane, by PhIO. The yield of oxygenation of heptane to heptanols and heptanones was found to increase from 38% when using Fe(TDCPP)Cl as catalyst to 78% with the third generation catalyst Fe(TDCPCl8P)Cl [143].
| heptane + PhIO → heptanols + heptanones + PhI | (27) |
Examples of porphyrin ligands of the third generation. H2(TDCPBr8P): Ar = 2,6-dichlorophenyl, R = Br; H2(TDCPCl8P): Ar = 2,6-dichlorophenyl, R = Cl; H2(TDCPNxP): Ar = 2,6-dichlorophenyl, R = xNO2+(8 − x)H; H2(TMPBr8P): Ar = 2,4,6-trimethylphenyl, R = Br; H2(TF5PF8P): Ar = pentafluorophenyl, R = F; H2(TF5PS4P): Ar = pentafluorophenyl, R = 4SO3H + 4H.
5.2.3 Choice of the oxygen atom source
Several oxygen atom donors were used in these biomimetic systems. The first ones, such as iodosylarenes, hypochlorites and amine N-oxides, only contain one oxygen atom and are well-adapted for selective and clean formation of metal–oxo intermediates. In that regard, satisfactory results were also obtained with peracids and potassium monopersulfate that involve a non-symmetrical O–O bond whose heterolytic cleavage is favored by the formation of good leaving groups such as carboxylic acids or sulfate. The situation was more complex with H2O2 or alkylhydroperoxides, because Fe(III)porphyrins, as many transition metal complexes, not only lead to the heterolytic cleavage of their O–O bond, as expected for the formation of (porphyrin cation radical) Fe(IV)O species, but also to an homolytic cleavage of this bond with formation of OH or OR intermediates. The addition of cocatalysts such as imidazole or pyridine derivatives in the presence or in the absence of carboxylic acids, or ammonium acetate, led to a spectacular improvement of the oxygenating properties of H2O2 or ROOH in the presence of catalytic amounts of Mn or Fe porphyrins. Actually, these cocatalysts were found to facilitate the heterolytic cleavage of the O–O bond of H2O2 or ROOH and the transfer of one of their oxygen atom to the metal. Thus, systems based on Mn(III)porphyrins of the second generation associated to such cocatalysts were found especially efficient for epoxidation of alkenes and hydroxylation of alkanes and electron-rich aromatic rings by H2O2, as illustrated by Eq. (28):
| (28) |
Many systems using O2 itself as the oxygen atom source and mimicking the P450 catalytic cycle of dioxygen activation have been reported. They were based on a Fe- or Mn-porphyrin catalyst and a reducing agent. First results showing that such systems are able to epoxidize alkenes and hydroxylate alkanes were published in 1979 by the group of Tabushi that used Mn(TPP)Cl as catalyst and NaBH4 as source of electrons [57a]. Other monooxygenase-like systems generally based on Mn(III)porphyrins of the first generation and less often on Fe(III)porphyrins, and various reducing agents have been published during the next 20 years. They used a great variety of electron donors such as ammonium borohydride, H2 in the presence of colloidal platinum, NADH analogs, ascorbate, Zn powder, Zn amalgam, and aldehydes. Electrochemical systems using a Mn-porphyrin catalyst, either in solution or attached to the electrode, were also reported to epoxidize alkenes and hydroxylate alkanes and aromatic compounds with O2 as a source of oxygen atom. In most cases, these systems suffered from an overconsumption of electrons on comparison to the ideal consumption of 2 electrons per mole of hydroxylated product of the monooxygenase equation. This is due to all the secondary, ‘decoupling’ reactions that occur at the level of the Fe(II)O2, Fe(III)OOH and FeO intermediates of the monooxygenase catalytic cycle (see Fig. 3). These reactions are minimized in well-coupled P450-dependent monooxygenases thanks to a sophisticated control of the electron and proton transfers by the protein. This control is much less efficient in model systems and has to be greatly improved in the future. However, it is noteworthy that the use of bases, such as imidazole, and/or acylating agents, such as carboxylic acid chlorides or anhydrides, which could facilitate the formation of high-valent metal–oxo species, has generally improved the efficacy of the Mn porphyrin/reductant/O2 systems (for a review on all these points, see Ref. [142]).
Another system based on a Ru porphyrin, Ru(TMP)(O)2, was found to catalyze the epoxidation of olefins by O2 without the need of a reductant. First results about this system were published in 1984 by Groves et al. [145]. Its mechanism of dioxygen activation greatly differed from that of P450, except that the active oxygen species responsible for alkene epoxidation also seems to be a metal–oxo complex. Its interest is to transfer the two oxygen atoms of O2 to the alkene substrate, without any consumption of a reducing agent (for a recent review on the use of Ru porphyrins in oxidative catalysis see Ref. [22]).
5.2.4 Development of supported metalloporphyrin catalysts
The development of supported metalloporphyrin oxygenation catalysts began in the mid-1980s, after a first report by Nolte and Drenth in 1983 about the epoxidation of alkenes by NaOCl catalyzed by a Mn porphyrin bound to a polyisocyanide polymer in the presence of 4-methylpyridine [146]. Then, a great variety of methods have been used to obtain supported Fe- or Mn-porphyrin catalysts that were used with some of the oxygen atom donors mentioned in Section 5.2.3 (mainly ClO−, PhIO, KHSO5 and ROOH). Thus, Fe, Mn and Ru porphyrins have been covalently bound to polystyrene or polyethylene glycol polymers, as well as to polypeptides. Negatively charged (sulfonated) Fe or Mn porphyrins were strongly adsorbed on ion-exchange resins bearing positive charges such as poly(vinylstyrene) polymers containing ammonium groups or poly(vinylpyridinium) polymers crosslinked with divinylbenzene. Similar strong electrostatic interactions have been used to prepare supported catalysts from adsorption of Fe and Mn porphyrins bearing positive charges, such as Fe and Mn complexes of meso-tetra-(4-methylpyridiniumyl)porphyrin, onto negatively charged ion-exchange resins such as Dowex. Adsorption or covalent binding of Fe and Mn porphyrins to mineral matrices such as silica, alumina or clays was also used for the construction of supported, catalytically active materials. Encapsulation of metalloporphyrins and metallophthalocyanins also led to supported oxidation catalysts. Another class of supported catalysts in which the Fe or Mn porphyrin is an integral part of the polymer was prepared either from the reaction of Fe(TF5PP)Cl with Na2S, which led to a metalloporphyrin-based polymer, or by co-condensation of Si(OEt)4 and iron porphyrins bearing SiF3 groups, which led to hybrid organic–inorganic oxidation catalysts. For review articles on Section 5.2.4 see Refs. [142,147].
5.2.5 Synthetic metalloporphyrins as regio- and enantioselective oxidation catalysts
Since the first report in 1983 on the use of a chiral iron porphyrin to catalyze the asymmetric epoxidation of p-chlorostyrene by PhIO (50% ee) [148], many articles have been published on metalloporphyrins as regio- and/or enantioselective catalysts. In these catalysts, special structural motifs have been attached to the porphyrin in order to achieve a selective recognition of the substrate and its specific positioning towards the metal–oxo active species. In P450s, this selective recognition and positioning of the substrate in the active site are due to specific interactions with protein residues that are close to the heme. For recent reviews on metalloporphyrin-catalyzed regio- and enantioselective oxidations, see Refs. [22,142,149–151]. The reader could also see recent reviews of Jacobsen and Katsuki on asymmetric epoxidation of alkenes catalyzed by chiral Schiff base complexes that are clearly related to metalloporphyrins [152,153].
5.3 Some applications of biomimetic metalloporphyrin oxidation catalysts
Sections 3 and 4 show how the use of iron porphyrins was important for understanding the nature of the stable intermediate iron complexes of the P450 catalytic cycle and of the various P450 iron–metabolite complexes spectrally detected upon metabolism of some substrates. Iron porphyrins were also very helpful to understand the nature and reactivity of the highly unstable intermediates of the P450 catalytic cycle (Fig. 3). This is the case of the Fe(III)OO− and Fe(III)OOH complexes that have been only detected by sophisticated EPR and ENDOR techniques at very low temperatures [21]. This is also the case of the perferryl iron–oxo species.
5.3.1 Use of iron porphyrins to mimic the Fe(III)OO−, Fe(III)OOH and iron–oxo intermediates of the P450 catalytic cycle
Reactions of ferric porphyrins with some of the oxygen atom donors indicated in Section 5.2.3 under carefully specified conditions have provided an access to various ferric superoxo, ferric peroxo and high-valent iron–oxo complexes. The great interest of these model studies, when compared to the corresponding studies using hemeproteins, is to easily change the nature of the porphyrin and the axial iron ligand to compare their effects, and to handle much more easily the resulting complexes for spectral studies under a great variety of conditions. Actually, these model studies have provided crucial data for understanding not only the unstable intermediates of the P450 catalytic cycle, but also the corresponding intermediates of other hemeproteins such as peroxidase, catalase, prostaglandin synthase or NO-synthase. Two detailed reviews have been published on the synthesis, spectral characterization, and reactivity of these porphyrin iron–oxo and –peroxo complexes [154,155].
First data in this field were published by Groves et al. in 1981. They showed the formation of a green species by the reaction of Fe(TMP)Cl with meta-chloroperbenzoic acid at −78 °C, which exhibited spectral characteristics analogous to those of peroxidase compound I [156]. An oxo–ferryl porphyrin Π-cation radical structure was proposed for this green species [156] (Eq. (29)).
| (29) |
Further studies on the reactions of iron(III)porphyrins with various oxygen atom donors such as peracids, iodosylarenes or O3, at low temperature, showed the formation of numerous (porphyrin cation radical) Fe(IV)O and (porphyrin)Fe(IV)O complexes. The detailed UV–vis, 1H NMR, EPR, Mössbauer and resonance Raman characteristics of these complexes were compared in Ref. [154], as a function of the porphyrin and axial ligands of iron. Their electronic structure and reactivity towards substrates were also discussed by comparing the corresponding hemeprotein iron–oxo complexes in recent review articles [22,154,155].
Ferric hydroperoxo, Fe(III)–OOH, species have been produced by the reaction of H2O2 with iron porphyrins and non-heme iron complexes under carefully controlled conditions, and their spectroscopic properties have been compared to those of the Fe(III)OOH intermediate of the P450 catalytic cycle (Fig. 3) and of other hemeprotein Fe(III)OOH species. The latter species were generated by cryoradiolytic reduction of the corresponding hemeprotein Fe(II)O2 complexes. This method was very important as it gave a first access to some spectral characteristics of hemeprotein Fe(III)OO− and Fe(III)OOH species (see Ref. [20] for a review on this point). Their EPR spectra were found to be sensitive to the protonation state of the peroxo ligand, allowing a clear differentiation between Fe(III)OOH and Fe(III)OO− species [21].
Porphyrin ferric peroxo complexes were prepared for the first time in 1980 by reaction of superoxide with Fe(III)porphyrins according to Eq. (30) [157].
| (30) |
These complexes, in which the O2− ligand appeared to be bound in a bidendate triangular fashion, were found to be unable to epoxidize tetramethylethylene or to hydroxylate alkanes. More recently, the peroxo complexes of electron-rich iron porphyrins such as Fe(TMP) or Fe(PPIXDME) were found to be able to epoxidize electron-poor alkenes such as cyclohexenone and menadione. These porphyrin iron peroxo complexes also reacted with aldehydes to yield deformylated products. These reactions were interpreted on the basis of a nucleophilic reactivity of the peroxo complexes (see Refs. [158,159], and references cited therein for these peroxo complexes). The mechanism proposed for the oxidative deformylation of aldehydes by the porphyrin iron peroxo complexes is shown in Fig. 10. It is very similar to the one generally admitted for the corresponding reactions catalyzed by P450s, which play a key role in oxidative demethylations occurring in steroid biosynthesis [9]. Such a nucleophilic attack of an intermediate ferric peroxo species is proposed to occur during the oxidation of Nω-hydroxy-l-arginine to l-citrulline and NO by NO-synthases [10,11,155,160] (Fig. 10). As far as the formation of a ferric peroxo complex of NO-synthase is concerned, very recent data have shown that the reaction of a stable porphyrin Fe(II)O2 complex with tetrahydrobiopterin, a reducing cofactor present in the NO-synthase active site, at low temperature, led to a transient ferric hydroperoxo species that was characterized by EPR spectroscopy [161].

Comparison of the mechanisms of P450-catalyzed oxidative deformylation of aldehydes and NO-synthase-catalyzed oxidation of Nω-hydroxyarginine to citrulline and NO. In the second reaction, R = NH(CH2)3CH(NH2)COOH. Non-concerted evolutions of the Fe(III)–OOX intermediates have also been proposed.
5.3.2 Use of biomimetic metalloporphyrin systems for the preparation of drug metabolites
Metalloporphyrins and related Schiff base complexes have been extensively used for selective oxygenations of a great variety of substrates in fine chemistry, for the oxidative degradation of pollutants that are resistant towards microbial destruction, such as polycyclic aromatic hydrocarbons or halogenated pesticides, and for selective DNA oxidative cleavage (for a review see Ref. [142]). Considering the key role of P450s in drug metabolism and the great importance of the knowledge of drug metabolism in the research and development of new therapeutic agents, a very helpful practical application of biomimetic oxidation catalysts is the preparation of drug metabolites. A direct access to these metabolites from a one-step oxidation of the parent drug is important (i) to determine their structure, (ii) to test their potential pharmacological effects and/or their toxic effects as soon as possible, and (iii) to patent them. Usually, the oxidation of a drug by the human P450s that are present mainly in the liver and the gastro-intestinal system leads to several metabolites coming from the oxidation of different regions of the molecule. Each human P450 leads to a more or less regioselective oxidation of the drug as a function of its mode of recognition and positioning of this drug relative to the heme by some protein residues of the active site. In most metalloporphyrin-based systems, such a specific positioning of the substrate does not exist. Therefore, the regioselectivity of its oxidation is dictated by the intrinsic chemical reactivity of its different functions. Generally, model systems led to a mixture of oxidized metabolites, which could be interesting to obtain several of these metabolites in one step. The theme of Section 5.3.2 has been the subject, among others, of a review article [142]. Examples of the use of metalloporphyrins for the oxidation of drugs are given as an illustration of their interest in this particular application. First examples were reported in the late 1980s, with the oxidation of antergan to N-dealkylated metabolites [162] and of phencyclidine to a piperidine-3-oxo metabolite [163] by the simple PhIO/Fe(TPP)Cl system, and with the regioselective hydroxylation of the thiophene ring of tienilic acid and suprofen by C6F5IO in the presence of Mn(TDCPP)Cl [102] (Eq. (31)).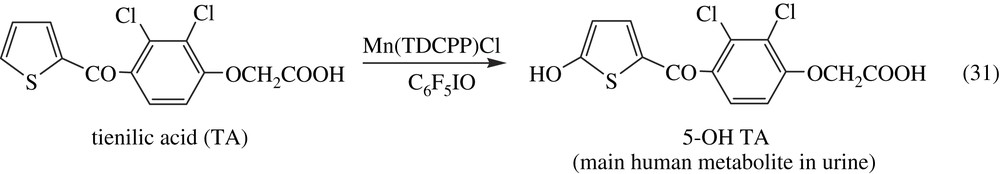
Later, it was found that the use of several biomimetic systems derived from variation of the nature of the metalloporphyrin catalyst, the oxygen atom donor or the reaction conditions, gave access to the different oxidized in vivo metabolites of drugs such as aminopyrine, lidocaine [164,165] (Fig. 11), propiverin [166,167], clemastin [168] and SR48117 [169]. Oxidation of antiulcer thioacetamide compounds upon oxygen activation on an electrode bearing polypyrrole–Mn-porphyrins gave access to some oxidized metabolites [170]. This suggests that proper electrochemical systems might be used to prepare oxidized metabolites of drugs.
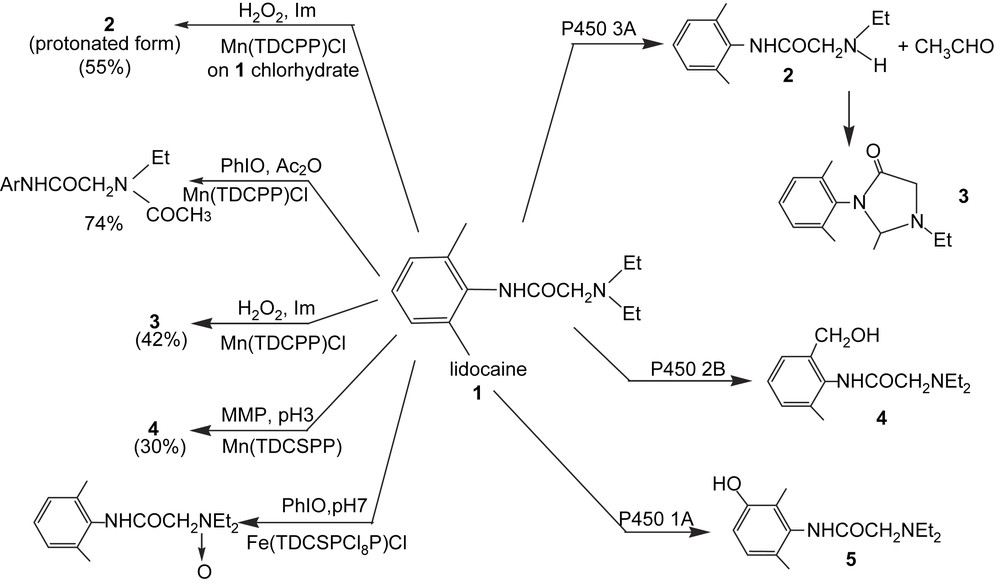
Different metabolites of lidocaine that may be prepared by using metalloporphyrin catalysts under various conditions. For the P450-dependent reactions, see Ref. [164]; for the metalloporphyrin-catalyzed reactions see Refs. [164,165]. Formula and abbreviations of the metalloporphyrins in Figs. 1 and 9 except for TDCSPP which means meso-tetra-(2,6-dichloro-3-sulfonatophenyl)porphyrin; MMP = magnesium monoperphthalate.
Data comparing the oxidation of the anti-inflammatory drug diclofenac, either by metalloporphyrin-based systems, or by human P450s expressed in yeast under a catalytically active form, have shown how these systems may be complementary for drug metabolites synthesis [171] (Fig. 12).

Metalloporphyrin catalysts and yeast-expressing human P450s as complementary tools for the preparation of metabolites of the drug diclofenac (from Ref. [171]).
Finally, biomimetic systems were used for the preparation of reactive metabolites of drugs. These reactive, generally electrophilic metabolites, that are formed upon oxidation of several drugs by P450s, covalently bind to cell macromolecules such as proteins or nucleic acids. This is at the origin of many secondary toxic effects of drugs, and must be predicted at an early stage of drug research. These reactive metabolites usually cannot be prepared upon oxidation of drugs by in vitro systems containing P450s such as liver microsomes because they too rapidly bind, in an irreversible manner, to the proteins and other nucleophiles present in the system. The situation is different with a biomimetic system in which one can avoid the presence of too reactive nucleophiles. Thus quinone–imine metabolites of acetaminophen or ellipticine [172,173] were obtained by biomimetic oxidations of the parent compound.
5.4 Catalytic iron porphyrin models for P450 reactions not consuming O2
The use of iron porphyrin catalysts was also important for understanding P450-dependent reactions that do not consume O2. Among these reactions, such as isomerizations, dehydrations or reductions (for reviews see [10,11,12,174]), the ones performed by the ferrous state of the P450 catalytic cycle have been mimicked by using iron porphyrins. Thus, the reduction of various xenobiotics, which may be catalyzed by P450 Fe(II), especially under anaerobic conditions, was performed by chemical systems using a reducing agent in the presence of an iron porphyrin catalyst. For instance, a biphasic (H2O–toluene) system using sodium ascorbate as a reducing agent and Fe(TPP)Cl as a catalyst, in the presence of a phase transfer agent, was found to quantitatively reduce hexachloroethane to tetrachloroethylene, phenanthrene-9,10-oxide to phenanthrene, and nitroso- or nitrobenzene to aniline [175].
P450-dependent dehydration of aldoximes is involved in the biosynthesis of cyanogenic glucosides from tyrosine in plants [176]. It was also reported in 1992 that liver P450s catalyzed the dehydration of butyraldoxime to butyronitrile [177]. This reaction was found to be quite general with alkyl- and arylaldoximes and a P450 Fe(II)–aldoxime complex, characterized by a Soret peak at 442 nm, appeared to be an intermediate in aldoxime dehydration [178]. Chemical model systems, based on iron porphyrin catalysts in the presence of a reducing agent to maintain their Fe(II) state and on a mild acid such as CH3COOH, also dehydrated aldoximes to nitriles in an efficient manner [179] (Eq. (32)). These model studies led to the isolation and characterization of porphyrin Fe(II)–aldoxime complexes, that correspond to the 442 nm absorbing P450 Fe(II)–aldoxime complexes. They showed that aldoximes were bound to Fe(II) via their nitrogen atom in the porphyrin Fe(II)–aldoxime complexes, and that the best catalysts for aldoxime dehydration were electron-rich iron porphyrins bearing an electron-donating axial ligand. These data suggested a mechanism for P450- or iron porphyrin-catalyzed dehydration of aldoximes involving an activation of the oxime function in Fe(II)–aldoxime complexes due to a partial charge transfer from Fe(II) to the CN–OH function. This would lead to the appearance of a partial negative charge on the oxime carbon atom that facilitates the β-elimination of its OH group presumably assisted by an acid catalyst. In the resulting cationic intermediate, the hydrogen present on the oxime carbon is highly acidic because of its β-position relative to iron, and should be easily removed in a fast step (Fig. 13).
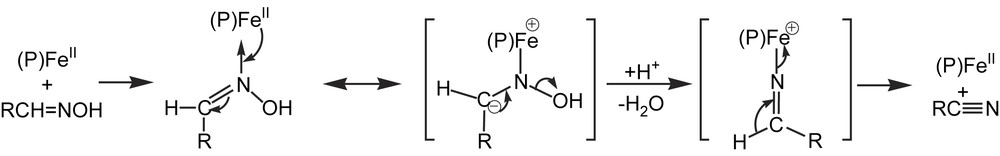
Possible mechanism for the P450- and iron porphyrin-catalyzed dehydration of aldoximes (from Refs. [178,179]).
6 Conclusion
The huge amount of studies that have been performed during these last 30 years on chemical models based on metalloporphyrins has led to several important contributions to the fields of cytochrome P450 and oxidation catalysis. This is illustrated in Fig. 14 which schematically compares the history of the development of our knowledge on P450 chemistry and the use of metalloporphyrin models.

Schematic comparison of the history of the development of our knowledge on P450 chemistry and of the use of metalloporphyrin models.
Historically, the first contribution was, in the 1975–1980 period, the synthesis of well-defined, structural models of the first four intermediates of the P450 catalytic cycle. These data completely confirmed the presence of an axial thiolate ligand on P450 iron, that had been proposed on the basis of P450 spectral data, a long time before the publication of the first P450 X-ray structure.
The second main contribution concerned the determination of the nature of various stable P450 iron–metabolite complexes, thanks to the synthesis and complete characterization of the corresponding porphyrin iron–metabolite complexes. These studies led to the discovery of a very rich coordination chemistry and organometallic chemistry of iron porphyrins and P450s, with first evidences for the existence of σ-alkyl (or aryl), carbene, nitrene and nitrosoalkane iron complexes. In many cases, the formation of such complexes was later found to occur with many other hemeproteins such as hemoglobin, prostaglandin synthase and NO-synthase. In the case of the P450 iron–metabolite complexes that are very stable in vivo and involved in problems of drug interactions, the determination of the nature of the metabolites acting as strong ligands was of great importance to interpret such drug interactions at the molecular level and also to predict them. In the case of P450-iron–metabolite complexes whose dissociation of the iron–metabolite bond leads to reactive species, the knowledge of the metabolite structure and of the reactivity of the iron–metabolite complex was useful for interpreting the toxic effects of the parent compound.
The third main contribution of iron porphyrin models is related to the preparation of many reactive iron–peroxo, –hydroperoxo and –oxo complexes that correspond to the transient intermediates of the P450 catalytic cycle (Fig. 3). The study of these model complexes has provided very important data for understanding the electronic structure and reactivity of the corresponding P450 unstable intermediates. It is noteworthy that these data were also crucial for understanding the structure and reactivity of the corresponding complexes of many other hemeproteins involved in dioxygen binding and/or activation and in different oxidative reactions. They should also be interesting starting points for understanding the nature and reactivity of the corresponding oxo and peroxo complexes involved in the catalytic cycles of non-heme iron oxygenases.
Finally, the fourth main contribution of metalloporphyrin model studies was the development of a great variety of selective oxidation catalysts. Systems using robust Fe or Mn porphyrins, as homogeneous or supported catalysts, and readily available and simple oxygen atom donors such as H2O2 or ClO− are already able to epoxidize olefins, to hydroxylate alkanes and to transfer an oxygen atom to sulfur- and nitrogen-containing substrates with a great efficacy. They are presently used for the preparation of oxidized metabolites of drugs in the pharmaceutical industry. Such systems for selective oxygen atom transfer have been (see above) and will be a source of inspiration for the design of similar non-porphyrin metal catalysts.
Progress is still required to further improve these metalloporphyrin-based oxidation systems, mainly in two directions. Improvement of the selectivity of substrate recognition by the catalyst or the support of the catalyst, which is performed by the protein in P450s, should lead to more regio-, stereo- and enantioselective oxygenations. Major improvements are also necessary for the construction of efficient systems for substrate hydroxylation by O2 itself, because of the difficulty to mimic the catalytic cycle of O2 activation by P450 that involves a tight coupling between the electron- and proton-transfer steps.