

1 Introduction
L'activation des réactions demeure une préoccupation fondamentale des chimistes, puisque l'optimisation du rendement chimique est intimement liée au mode d'activation choisi.
À cet égard, on a développé, au cours de ces dernières années, des procédés de catalyse sélective à hauts rendements. Parmi les techniques de réactions hétérogènes sans solvant, on distingue:
- – les réactions sur supports solides [1–15] ;
- – la catalyse par transfert de phase solide/liquide en en l’absence de solvant [4,16–23] ;
- – les réactions sans solvant, ni catalyseur, ni support [10,24–26].
Dans ce travail, nous nous proposons d'examiner l'effet de l'activation anionique par catalyse par transfert de phase solide/liquide en l'absence de solvant, qui trouve particulièrement son domaine d'application dans les réactions anioniques.
Il s'agit là de réactions sans solvant ni support, mais catalysées par un agent de transfert de phase [18,19]. Cette technique, qui permet la réalisation de réactions virtuellement impossibles, sera décrite dans le cas des additions de Michael et de Baylis–Hillman.
Les résultats obtenus seront argumentés par les mécanismes réactionnels correspondants.
2 Résultats et discussion
2.1 Réaction de Baylis–Hillman
La condensation entre un aldéhyde et un composé acrylique catalysée par une amine tertiaire cyclique de type 1,4-diaza-2,2,2-bicyclooctane (DABCO) ou quiniclidine (QN) conduit à la formation de l'adduit de Baylis–Hillman. Dans un premier stade, nous avons étudié l'influence de la polarité du solvant sur le rendement de la réaction de Baylis–Hillman entre le benzaldéhyde et l'anhydride maléique (Tableau 1) :
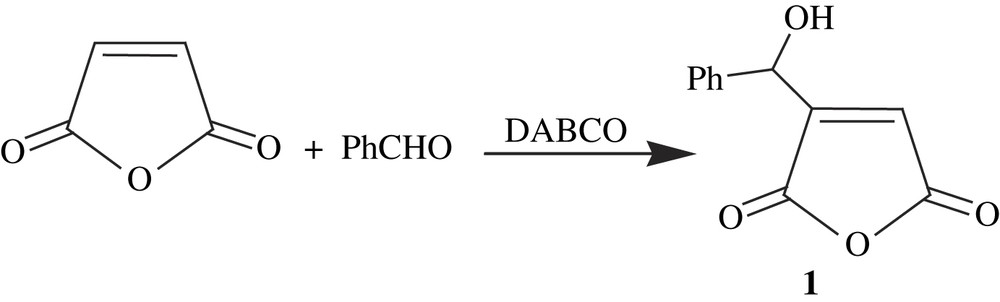
Influence de la polarité du solvant sur le rendement de la réaction de Baylis–Hillman entre le benzaldéhyde et l'anhydride maléique
| Solvant | ɛ à 25 °C | μ (10−30 cm−1) | Durée (h) | Rdt (%) |
| Toluène | 2.38 | 11,20 | 170 | 8 |
| THF | 7,58 | 5,90 | 150 | 11 |
| Chloroforme | 4,7 | 13,50 | 92 | 11 |
| DMF | 36,7 | 10,80 | 80 | 39 |
| Acétonitrile | 35,94 | 5,70 | 48 | 39 |
| Méthanol | 33 | 5,80 | 34 | 45 |
| Formamide | 109,5 | 3,80 | 10 | 59 |
| Eau | 87 | 0,00 | 10 | 67 |
L'étude de cette réaction dans une série de différents solvants organiques fait apparaître une grande dépendance de l'évolution du système vis-à-vis de la polarité du solvant. En revanche, les rendements obtenus tendent à diminuer et les durées à augmenter quand on passe d'un solvant polaire à un solvant apolaire. Pour une même durée, on constate que le rendement est plus important quand on opère en solution aqueuse que lorsqu'on travaille en présence de formamide. Ceci semble être dû à la stabilité de la forme zwitterionique par le biais des liaisons hydrogène [27]. Notons aussi que les rendements obtenus dans les différents milieux examinés ne sont pas quantitatifs, par comparaison avec la réaction entre le benzaldéhyde et l'acrylonitrile [16]. La raison principale en est probablement la faible solubilité de l'amine tertiaire cyclique de type DABCO dans les divers solvants. Dans ce contexte, Auge et Lubineau [28] ont montré l'intérêt de réaliser la réaction de B–H en milieu aqueux, où le taux de conversion atteint est de l'ordre de 90%. Dans un autre travail, mené par Lee et Gong, on montre aussi que la préparation des adduits de B–H impliquant des cycloalcénones est favorisée en milieu aqueux avec le DMAP (4-(diméthylamino)pyridine) comme catalyseur [29]. Ainsi, il été montré que le choix de l'eau comme milieu réactionnel accélérait la réaction de Baylis–Hillman [30–33], dans laquelle la condensation de l'acrylate de méthyle avec différents aldéhydes en présence de DABCO en quantité stœchiométrique fournit les adduits de B–H correspondants en des durées courtes [34]. Dans ces conditions, même les acrylamides, qui sont peu réactives, réagissent avec les aldéhydes pour former les adduits de B–H, avec une excellente réactivité [33].
Afin d'augmenter les rendements et d'améliorer les conditions opératoires en termes de durée et de température, nous avons soumis cette réaction à la catalyse par un agent de transfert de phase. Ainsi, la réaction de Baylis–Hillman entre le benzaldéhyde et l'anhydride maléique a eu lieu en quantités quasi-équivalentes, en présence d'une quantité catalytique de sel d'ammonium et dans différentes conditions opératoires. Le Tableau 2 illustre les résultats obtenus.
Influence de la CTP sur le rendement de la réaction de Baylis–Hillman entre le benzaldéhyde et l'anhydride maléique
| Produit | CTP | Milieu | Rdt% |
| 1 | Aucun | Eau + DABCO | 67 |
| CH2Cl2 + DABCO | 19 | ||
| (*) + DABCO | 5 | ||
| TBAB | Eau + DABCO | 69 | |
| CH2Cl2 + DABCO | 43 | ||
| (*) + DABCO | 56 | ||
| Aliquat 336 | Eau + DABCO | 74 | |
| CH2Cl2 + DABCO | 59 | ||
| (*) + DABCO | 67 |
En milieu aqueux et en présence de CTP, les liaisons hydrogène [27] engendrent une stabilité de la forme zwitterionique obtenue lors de l'addition d'amine tertiaire sur la double liaison activée de l'anhydride maléique. Dans ce cas, la réaction a eu lieu avec un rendement de 67%, ce qui n'est pas le cas en milieu organique, où il ne dépasse guère 19%. Par ailleurs, l'ajout de CTP n'affecte pas le rendement dans l'eau. En revanche, sa réactivité est nettement accrue dans le dichlorométhane, où les rendements sont améliorés. Ces résultats laissent prévaloir un effet positif de la CTP, en milieu organique davantage qu'en milieu aqueux.
En revanche, si nous comparons les rendements obtenus dans les divers milieux, nous constatons que l'Aliquat-336 semble être l'agent le plus adéquat pour la synthèse du produit de Baylis–Hillman [16]. Cet adduit est obtenu avec un rendement de 5% en l’absence de solvant et de CTP i ce résultat est prévisible, car l'anhydride maléique et le DABCO ne sont que partiellement solubles dans la phase liquide. De même, les rendements sont accrus en présence de CTP selon un processus de catalyse solide/liquide en l'absence de solvant (Schéma 1). On conçoit que l'attaque de la forme zwitterionique, obtenue lors de la première étape de la réaction, sur la double liaison CO de l'aldéhyde serait plus favorisée en présence de CTP. Ceci peut s'expliquer par la stabilité de la forme zwitterionique dans ces conditions, par réaction ionique avec le sel d'ammonium, comme le montre le mécanisme proposé de la réaction de Baylis–Hillman, impliquant le benzaldéhyde et l'anhydride maléique en catalyse hétérogène liquide–solide (Schéma 1). Cependant, en présence d'un solvant, la réaction se déroule selon un processus de catalyse homogène en phase liquide [30].

Mécanisme de la réaction de B–H entre le benzaldéhyde et l'anhydride maléique en présence de CTP : catalyse hétérogène liquide–solide.
2.2 Réaction de Michael
C'est une addition nucléophile sur la double liaison CC d'une cétone, d'un ester ou un nitrile α,β-insaturée. Cette réaction joue un rôle important dans le domaine de la synthèse organique [35,36]. Une variété d'amines réagit avec les composés α,β-éthyléniques pour donner les dérivés β-amino correspondants [37–39] :
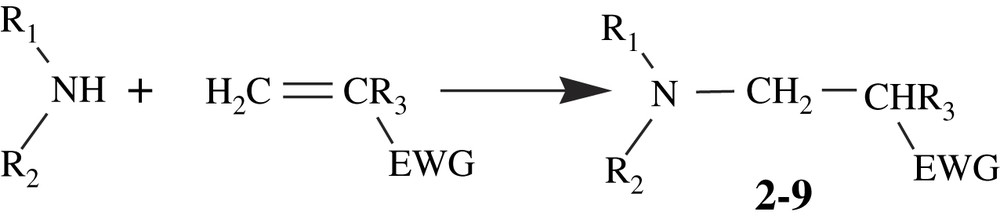
Les réactions de Michael impliquant des esters acryliques de type acrylate de méthyle ou méthacrylate de méthyle se distinguent par une absence de réactivité en milieu organique et en milieu aqueux, alors que, dans le cas d'une cétone ou nitrile α,β-insaturée, la réactivité est accrue en milieu aqueux (Tableau 3). Ceci s'explique par le fait que l'adduit de Michael est soumis, dans le cas des esters acryliques, à une réaction réversible de type rétro-Michael générant les réactifs de départ. En effet, la réaction de Michael débute par une addition nucléophile générant la liaison C–N et la formation d'un intermédiaire zwitterionique. En milieu aqueux ou dans le méthanol, cette forme zwitterionique est stabilisée par le biais de liaisons hydrogène, inhibant ainsi toute éventuelle décomposition de type rétro-Michael.

Influence du milieu sur le rendement de la réaction de Michael
| R1 | R2 | R3 | EWG | produit | Solvant | Rdt en % (durée) |
| Ph | H | H | CN | 2 | Aucun | 32 (4 h) |
| Acétonitrile | 0 (4 h) | |||||
| Méthanol | 5 (4 h) | |||||
| Eau | 37 (4 h) | |||||
| CO2CH3 | 3 | Aucun | 28 (4 h) | |||
| Acétonitrile | 0 (4 h) | |||||
| Méthanol | 0 (4 h) | |||||
| Eau | 0 (4 h) | |||||
| COCH3 | 4 | Aucun | 30 (4 h) | |||
| Acétonitrile | 0 (4 h) | |||||
| Méthanol | 8 (4 h) | |||||
| Eau | 26 (4 h) | |||||
| CH3 | CO2CH3 | 5 | Aucun | 17 (4 h) | ||
| Acétonitrile | 0 (4 h) | |||||
| Méthanol | 0 (4 h) | |||||
| Eau | 0 (4 h) | |||||
| Ph-CH2 | CH3 | H | CN | 6 | Aucun | 93 (1 h) |
| Acétonitrile | 0 (4 h) | |||||
| Méthanol | 15 (4 h) | |||||
| Eau | 85 (2 h) | |||||
| CO2CH3 | 7 | Aucun | 89 (2 h) | |||
| Acétonitrile | 0 (4 h) | |||||
| Méthanol | 0 (4 h) | |||||
| Eau | 0 (4 h) | |||||
| COCH3 | 8 | Aucun | 88 (2 h) | |||
| Acétonitrile | 0 (4 h) | |||||
| Méthanol | 17 (4 h) | |||||
| Eau | 78 (2 h) | |||||
| CH3 | CO2CH3 | 9 | Aucun | 84 (2 h) | ||
| Acétonitrile | 0 (4 h) | |||||
| Méthanol | 0 (4 h) | |||||
| Eau | 0 (4 h) |
Cependant, avec le méthanol comme solvant, on pourrait s'attendre à son addition sur la double liaison CC. Pourtant, cette réaction est très limitée dans nos conditions. En effet, l'analyse expérimentale n'a pas décelé la présence de produits issus de cette réaction. Ceci est en accord avec les travaux de Jenner [40,41], qui a montré que cette addition est favorisée sous une pression de 300 MPa.
Une comparaison des résultats obtenus avec les deux amines montre que la réactivité est meilleure avec le benzyl méthyl amine qu'avec l'aniline. En effet, la paire d'électrons libre portée par l'atome d'azote est responsable du caractère nucléophile des amines. Cette paire libre dans l'aniline se trouve délocalisée par interaction avec les orbitales π du noyau aromatique et, par conséquent, est moins disponible pour réagir comme agent nucléophile. En revanche, dans le benzyl méthyl amine, l'absence de cette délocalisation et la présence d'un groupe méthyle, donneur par effet inductif (+I), rend la paire d'électrons libre de l'azote plus disponible, pour que l'amine puisse réagir comme agent nucléophile.
En revanche, on montre l'intérêt de manipuler en l'absence de solvant, où la réaction se déroule avec d'excellents rendements et une grande sélectivité. Notons ici que la réaction nécessite un léger excès du composé acrylique.
L'addition de catalyseur à transfert de phase de type Aliquat-336 engendre une réactivité remarquable (Tableaux 3 et 4), dans la mesure où il permet d'améliorer le rendement et de minimiser le temps de réaction. Il semble que le sel d'ammonium permette, par réaction d'échange ionique, la stabilisation de la forme zwitterionique obtenue après attaque nucléophile de l'amine sur le composé acrylique et évite, par conséquent, toute éventuelle réaction de type rétro-Michael.
Influence d'une addition d'Aliquat-336 sur le rendement de la réaction de Michael
| R1 | R2 | R3 | EWG | Rdt en % (durée) |
| Ph | H | H | CN | 64 (2 h) |
| CO2CH3 | 62 (2 h) | |||
| COCH3 | 68 (2 h) | |||
| CH3 | CO2CH3 | 63 (2 h) | ||
| Ph-CH2 | CH3 | H | CN | 94 (0,5 h) |
| CO2CH3 | 91 (1 h) | |||
| COCH3 | 92 (1 h) | |||
| CH3 | CO2CH3 | 87 (1 h) |
Le produit 10 a été synthétisé par addition selon une réaction de Michael entre l'hexadécylamine et l'acrylonitrile :

La réaction a eu lieu en présence et en l'absence de solvant, mais avec des rendements relativement faibles. La faible réactivité en l'absence de solvant peut être expliquée par le fait que l'amine n'est pas totalement soluble dans la phase liquide. L'jout de CTP type Aliquat-336 augmente le rendement ; en l'absence de solvant, le rendement passe de 36 à 84 et la réaction se déroule selon un processus de catalyse solide–liquide, où l'acrylonitrile joue à la fois le rôle de solvant organique et de réactif électrophile (Schéma 2 et Tableau 5).
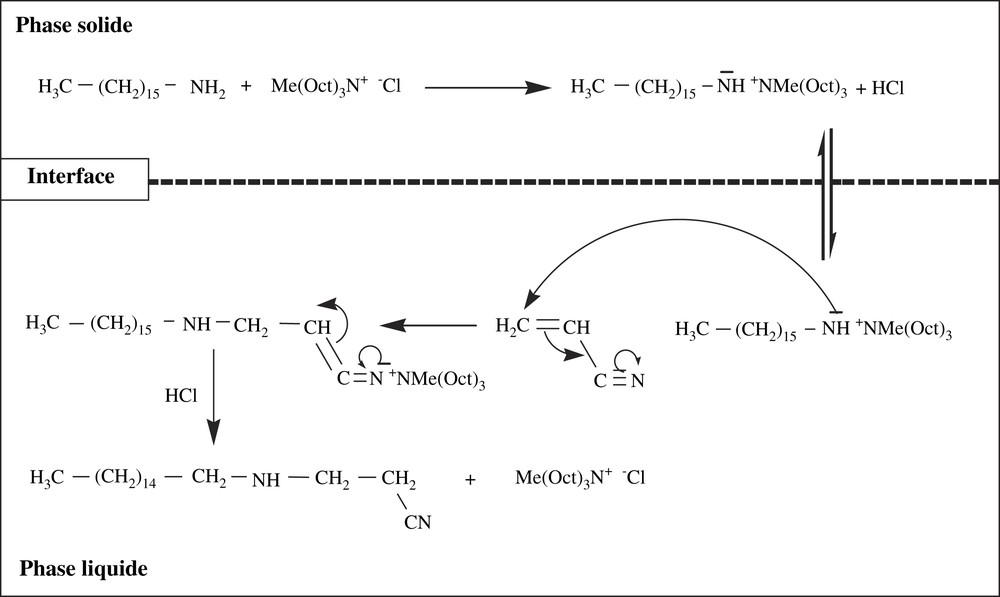
Mécanisme de la réaction de Michael entre l'hexadécylamine et l'acrylonitrile en présence d'Aliquat-336 : catalyse solide/liquide en l'absence de solvant.
Influence du milieu sur le rendement de la réaction de Michael entre l'hexadécylamine et l'acrylonitrile
| Sans CTP | Avec CTP | |||
| Milieu | Sans solvant | Avec solvant | Avec solvant | Sans solvant |
| Rdt (%) | 36 | 21 | 44 | 84 |
3 Conclusion
L'exaltation de la nucléophilie a été mise en évidence par l'échange d'ions avec des sels d'ammonium quaternaires. À cet égard, l'ajout d'un catalyseur de transfert de phase dans le milieu réactionnel favorise l'accroissement du rendement de la réaction de Baylis–Hillman entre le benzaldéhyde et l'anhydride maléique selon un processus de catalyse hétérogène solide–liquide en l'absence de solvant, selon un processus de catalyse homogène en présence de solvant. Par ailleurs, la réaction de Michael se comporte différemment vis-à-vis de la catalyse par transfert de phase en présence et en l'absence de solvant. Cependant, on note l'intérêt de manipuler en milieu aqueux pour la réaction de Baylis–Hillman et pour les réactions de Michael impliquant une cétone ou un nitrile α,β-insaturé(e). En l'absence de solvant, les réactions de Michael examinées dans ce travail sont largement favorisées.
4 Partie expérimentale
4.1 Mode opératoire
4.1.1 Réaction de Baylis–Hillman
Un mélange de 2,12 g (20 mmol) de benzaldéhyde, 1,96 g (20 mmol) d'anhydride maléique et 0,224 g (2 mmol) de DABCO dans 5 ml de solvant est agité pendant le temps désiré. Le mélange est ensuite dilué dans l'éther diéthylique (25 ml). On procède à une extraction avec une solution d'acide chlorhydrique 2 N, puis avec une solution de bicarbonate de sodium. La phase organique est séchée à l'aide de sulfate de magnésium anhydre. Le solvant est évaporé, le produit est purifié par chromatographie sur colonne de gel de silice en utilisant un mélange (hexane:éther = 95:5) comme éluant.
Le catalyseur à transfert de phase est additionné à raison de 10% (soit 2 mmol).
4.1.2 Réaction de Michael
Dans un ballon de 25 ml, équipé d'un agitateur magnétique, on introduit 726 mg (6 mmol) de benzylamine et 350 mg (5 mmol) de méthylvinylcétone. Le mélange est soumis à une agitation magnétique pendant 2 h à la température ambiante (20–23 °C). Après réaction, on réalise une chromatographie sur colonne de gel de silice en utilisant un mélange (hexane:éther = 3:7) comme éluant. L'adduit de Michael (4-(N-benzyl, N-méthylamino)butan-2-one) est obtenu pur, avec un rendement de 88% (840 mg).
Pour les réactions réalisées dans un solvant, on ajoute au mélange des deux réactifs 10 ml de solvant.
L'agent catalyseur de transfert de phase est utilisé à raison de 10% (soit 0,5 mmol).
4.2 Enregistrement des spectres
Les spectres de RMN du 1H (300 MHz) et 13C (75 MHz) ont été enregistrés sur un spectromètre Bruker AC300 dans le CdCl3, avec le tétraméthylsilane comme référence interne. Les multiplicités des signaux sont indiquées par les abréviations suivantes : s, singulet ; d, doublet ; t, triplet ; b, bonde ; m, multiplet ; d.d, doublet dédoublé.
Les spectres IR ont été réalisés dans KBr sur un spectromètre JASCO FT-IR-420, dont la précision est de ±2 cm−1 dans le domaine 4000–400 cm−1.
4.2.1 3-(Hydroxy-phényl-méthyl)furan-2,5-dione 1
IR : νCC = 1585 cm−1 ; νCO = 1710 cm−1 ; νOH = 3460 cm−1.
RMN 1H : (300 MHz) (CDCl3) : 3,82 (b, 1H) ; 5,56 (s, 1H) ; 6,74 (s, 1H) ; 7,22–7,38 (m, 5H).
RMN 13C : (75 MHz) (CDCl3) : 74,51 ; 127,45 ; 128,28 ; 129,79 ; 128,52 ; 141,11 ; 147,25 ; 164,49.
4.2.2 3-(N-Phénylamino)propionitrile 2 [39]
IR : νNH = 3340 cm−1 ; νCN = 2276 cm−1.
RMN 1H (300 MHz, CDCl3) : 2,47 (t, 2H, 3JHH = 6,9 Hz) ; 2,71 (t, 2H, 3JHH = 6,9 Hz) ; 3,77 (b, 1H) ; 6,62 (d, 2H, 3JHH = 7,8 Hz) ; 6,74 (d.d, 2H, 3JHH = 7,5, 7,8 Hz) ; 7,20 (t, 1H, 3JHH = 7,5 Hz).
RMN 13C (75 MHz, CDCl3) : 17,63 ; 51,36 ; 113,13 ; 117,56 ; 118,79 ; 129,52 ; 146,97.
4.2.3 3-(N-Phénylamino)propionate de méthyle 3 [39]
IR : νNH = 3342 cm−1 ; νCO = 1674 cm−1.
RMN 1H (300 MHz, CDCl3) : 2,76 (t, 2H, 3JHH = 6,9 Hz) ; 3,44 (t, 2H, 3JHH = 6,9 Hz) ; 3,69 (s, 3H) ; 3,78 (b, 1H) ; 6,63 (d, 2H, 3JHH = 7,8 Hz) ; 6,75 (d.d, 2H, 3JHH = 7,5, 7,8 Hz) ; 7,19 (t, 1H, 3JHH = 7,5 Hz).
RMN 13C (75 MHz, CDCl3) : 32,42 ; 40,34 ; 52,56 ; 113,82 ; 118,61 ; 129,84 ; 147,44 ; 173,28.
4.2.4 4-(N-Phénylamino)butan-2-one 4
IR : νNH = 3338 cm−1 ; νCO = 1675 cm−1.
RMN 1H (300 MHz, CDCl3) : 2,17 (s, 3H) ; 2,75 (t, 2H, 3JHH = 6 Hz) ; 3,42 (t, 2H, 3JHH = 6 Hz) ; 3,77 (b, 1H) ; 6,62 (d, 2H, 3JHH = 7,8 Hz) ; 6,75 (d.d, 2H, 3JHH = 7,5, 7,8 Hz) ; 7,20 (t, 1H, 3JHH = 7,5 Hz).
RMN 13C (75 MHz, CDCl3) : 30,41 ; 38,36 ; 42,62 ; 113,08 ; 117,69 ; 129,41 ; 147,74 ; 208,32.
4.2.5 2-Méthyl, 3-(N-phénylamino)propionate de méthyle 5
IR : νNH = 3344 cm−1 ; νCO = 1678 cm−1.
RMN 1H (300 MHz, CDCl3) : 1,34 (d, 2H, 3JHH = 7,2 Hz) ; 2,67–2,84 (m, 1H) ; 3,47 (d, 2H, 3JHH = 6,9 Hz) ; 3,68 (s, 3H) ; 3,79 (b, 1H) ; 6,62 (d, 2H, 3JHH = 7,8 Hz) ; 6,74 (d.d, 2H, 3JHH = 7,5, 7,8 Hz) ; 7,20 (t, 1H, 3JHH = 7,5 Hz). RMN 13C (75 MHz, CDCl3) : 14,60 ; 38,34 ; 51,62 ; 53,06 ; 114,02 ; 118,91 ; 129,87 ; 147,54 ; 173,17.
4.2.6 3-(N-Benzyl, N-méthylamino)propionitrile 6
IR : νCN = 2282 cm−1.
RMN 1H (300 MHz, CDCl3) : 2,28 (s, 3H); 2,49 (t, 2H, 3JHH = 6,9 Hz) ; 2,72 (t, 2H, 3JHH = 6,9 Hz) ; 3,56 (s, 2H) ; 7,27–7,35 (m, 5H).
RMN 13C (75 MHz, CDCl3) : 16,36 ; 41,84 ; 52,20 ; 61,90 ; 118,97 ; 127,42 ; 128,50 ; 128,93 ; 138,22.
4.2.7 3-(N-Benzyl, N-méthylamino)propionate de méthyle 7
IR : νCO = 1676 cm−1.
RMN 1H (300 MHz, CDCl3) : 2,21 (s, 3H) ; 2,53 (t, 2H, 3JHH = 7,2 Hz) ; 2,74 (t, 2H, 3JHH = 7,2 Hz) ; 3,51 (s, 2H) ; 3,67 (s, 3H) ; 7,24–7,34 (m, 5H).
RMN 13C (75 MHz, CDCl3) : 32,70 ; 41,93 ; 51,65 ; 52,70 ; 62,12 ; 127,12 ; 128,29 ; 129,03 ; 138,73 ; 173,08.
4.2.8 4-(N-Benzyl, N-méthylamino)butan-2-one 8
IR : νCO = 1678 cm−1.
RMN 1H (300 MHz, CDCl3) : 2,12 (s, 3H) ; 2,17 (s, 3H) ; 2,62 (d.d, 2H, 3JHH = 6, 6,6 Hz) ; 2,68 (d.d, 2H, 3JHH = 6, 6,6 Hz) ; 3,47 (s, 2H) ; 7,23–7,31 (m, 5H).
RMN 13C (75 MHz, CDCl3) : 30,03 ; 41,97 ; 42,06 ; 52,02 ; 62,38 ; 127,14 ; 128,32 ; 129,08 ; 138,74 ; 208,29.
4.2.9 2-Méthyl, 3-(N-benzyl, N-méthylamino)propionate de méthyle 9
IR : νCO = 1675 cm−1.
RMN 1H (300 MHz, CDCl3) : 1,32 (d, 3H, 3JHH = 7,2 Hz) ; 2,30 (s, 3H) ; 2,62–2,78 (m, 3H) ; 3,56 (s, 2H) ; 3,68 (s, 3H) ; 7,20–7,36 (m, 5H).
RMN 13C (75 MHz, CDCl3) : 15,36 ; 33,73 ; 42,03 ; 52,58 ; 54,72 ; 62,30 ; 127,15 ; 128,31 ; 129,11 ; 138,74 ; 173,06.
4.2.10 3-(N-Hexadécylamino)propionitrile 10
IR : νNH = 3345 cm−1.
RMN 1H (300 MHz, CDCl3) : 0,90 (t, 3H, 3JHH = 6,9 Hz) ; 1,25 (m, 28H) ; 2,52 (t, 2H, 3JHH = 6,6 Hz) ; 2,62 (t, 2H, 3JHH = 7,2 Hz) ; 2,93 (t, 2H, 3JHH = 6,6 Hz) ; 3,50 (b, 1H).
RMN 13C (75 MHz, CDCl3) : 14,14–31,94 ; 45,10 ; 49,28 ; 118,76.