

1 Introduction
Une oxydation sélective correspond à l'abstraction d'un ou de plusieurs atomes de H de l'hydrocarbure, à l'insertion d'un ou de plusieurs atomes d'oxygène dans la lacune et au transfert de plusieurs électrons. Elle fait intervenir simultanément les propriétés acido-basiques et redox du catalyseur.
L'oxydation des hydrocarbures par l'oxygène moléculaire sur des oxydes métalliques fait intervenir le plus souvent le mécanisme dit de Mars et Van Krevelen [1], dans lequel la molécule d'hydrocarbure est activée sur une site cationique, avec formation d'un radical allyle, puis arrachement d'une deuxième hydrogène et insertion d'un atome d'oxygène du réseau dans la molécule organique. Il y a donc réduction du catalyseur et ré-oxydation par l'oxygène moléculaire de la phase gazeuse. Dans les réactions d'oxydation des alcanes sur les hétéropolyanions de type Keggin, il a été montré que les atomes d'oxygène du réseau cristallin participent à l'acte catalytique [2,3]. Il est par ailleurs admis que ce sont les espèces oxygène nucléophiles (O2−) de la surface des oxydes qui favorisent les réactions d'oxydation sélective sans rupture de la liaison C–C, alors que les espèces oxygène électrophiles (O2−, O−) adsorbées à la surface du catalyseur sont responsables de la rupture de la liaison C–C et ainsi de la formation des COx [4–6].
Dans la réaction de déshydrogénation oxydante du cyclohexane, certaines études s'accordent à dire que le cyclohexène et le benzène sont formés par des réactions successives, selon le formalisme de Mars et Van Krevelin [7–9] : cyclohexane → cyclohexène → benzène.
Pour Akimoto et al. [10–12], l’étape limitante serait la réduction du catalyseur par le réactif et, pour T. Harberle et al. [13], ce serait la ré-oxydation du solide qui serait l’étape limitante. La constante de réduction déterminée, dans ce cas, est supérieure à celle de la ré-oxydation.
Dans des études précédentes [14–16], nous avons montré que les polyoxométallates phosphomolybdique et phosphovanadomolybdique de type Keggin, de formules Cs2,5M0,08H0,26PMo12O40 et Cs2,5M0,08H1,26PMo11VO40 (M: Ni, Fe), sont actifs et sélectifs dans l'oxydéshydrogénation du cyclohexane en benzène à 350 °C. Dans le présent travail, nous reportons les études cinétique et mécanistique de cette réaction sur des sels mixtes de césium et de fer : Cs2,5Fe0,08H0,26PMo12O40 (CsFePMo12) et Cs2,5Fe0,08H1,26PMo11VO40 (CsFePMo11V).
2 Partie expérimentale
Les hétéropolyacides H3PMo12O40 et H4PMo11VO40 ont été préparés selon la méthode classique [17]. Les sels ont été précipités par addition d'une solution aqueuse de Cs2CO3 à un mélange de H3PMo12O40 ou H4PMo11VO40 et Fe(NO3)3. Le solide formé est séché sous vide à 313 K pendant 5 h.
La réaction de déshydrogénation du cyclohexane a été réalisée à 350 °C dans un réacteur en quartz à lit fixe et sous pression atmosphérique. Le solide (200 mg) est prétraité sous oxygène à la température de réaction pendant 1 h. Le mélange réactionnel est obtenu en faisant passer le mélange gazeux (N2 + O2) à travers un saturateur contenant le cyclohexane. Ce dernier est maintenu à 7 °C, avec une pression partielle de 40 torr. Le débit total et le rapport N2/O2 sont de 3 l h−1 et 1 respectivement. Les réactifs et produits de la réaction (cyclohexane, benzène, cyclohexène, cyclohexadiène, n-hexane, méthane, O2 et CO) sont analysés par chromatographie en phase gazeuse (FID-TCD) de type Shimadzu 14A. La colonne utilisée est une Carbowax 20M.
Les études cinétiques ont été conduites à faible taux de conversion du cyclohexane, compris entre 5 et 10%. Pour déterminer l'ordre par rapport au cyclohexane, le pourcentage en oxygène a été fixé à 50% et celui en cyclohexane est compris entre 2,5 et 25%. Pour déterminer l'ordre par rapport à l'oxygène, le pourcentage de cyclohexane a été fixé à 5% et celui en oxygène est compris entre 5 et 10%. Le temps de contact est maintenu constant.
3 Résultats et discussion
3.1 Étude cinétique
3.1.1 Ordres réactionnels
Les résultats de l’étude des variations de vitesse en fonction des pressions partielles en cyclohexane et en oxygène moléculaire (à pressions constantes en oxygène moléculaire et cyclohexane, respectivement) sont rapportés dans les Figs. 1 et 2. Il ressort de cette étude que la vitesse de la réaction augmente avec la pression partielle en cyclohexane (Fig. 1), mais qu'elle est indépendante de celle de l'oxygène moléculaire (Fig. 2). Si α et β sont les ordres réactionnels par rapport au cyclohexane et à l'oxygène moléculaire, la vitesse de la réaction s’écrit :
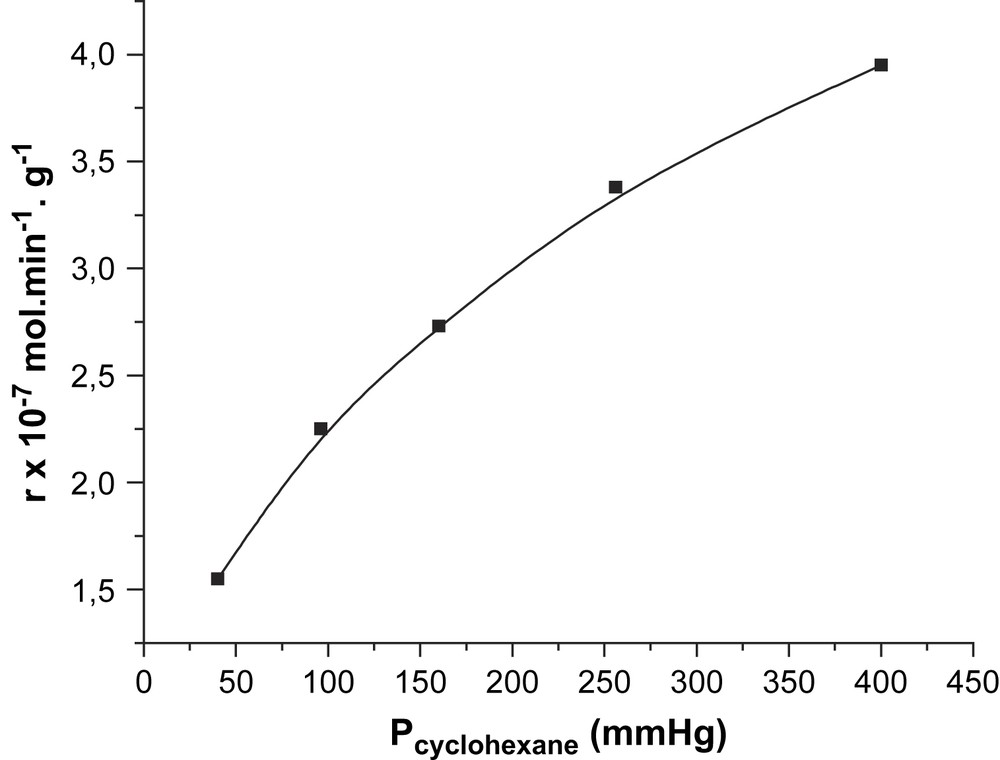
Variation de la vitesse en fonction de la pression partielle de cyclohexane.
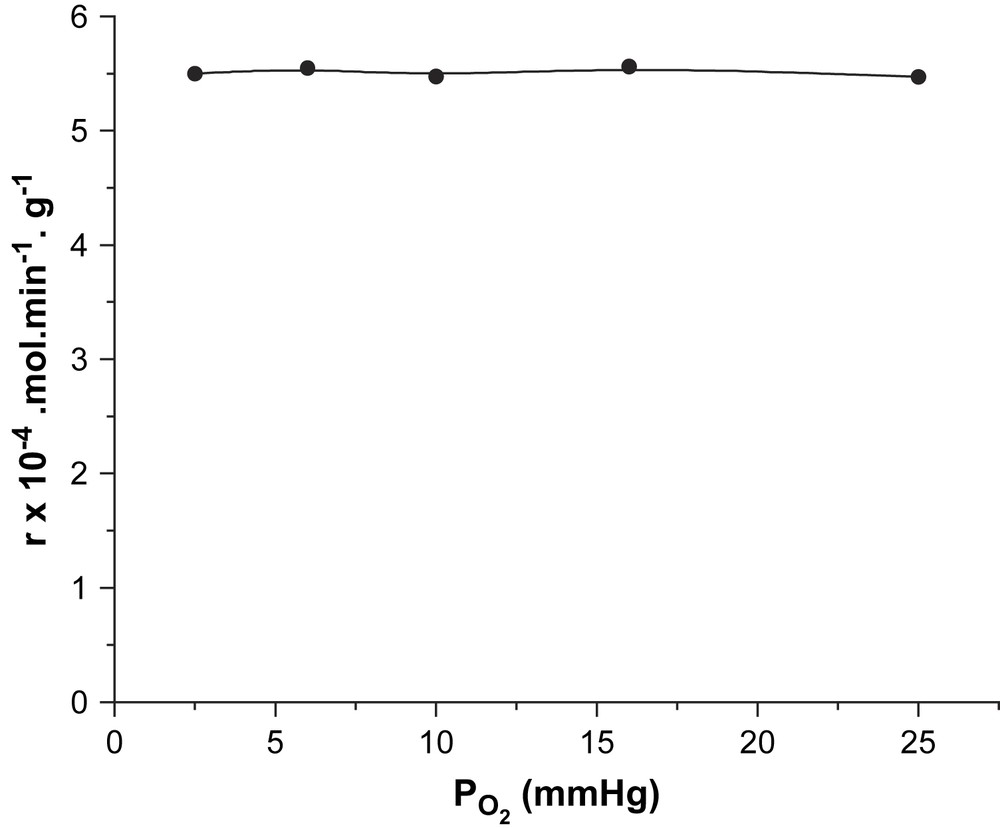
Variation de la vitesse en fonction de la pression partielle de l'oxygène.
Les valeurs de ces ordres, obtenues en traçant les courbes logarithmiques
| (1) |
| (2) |
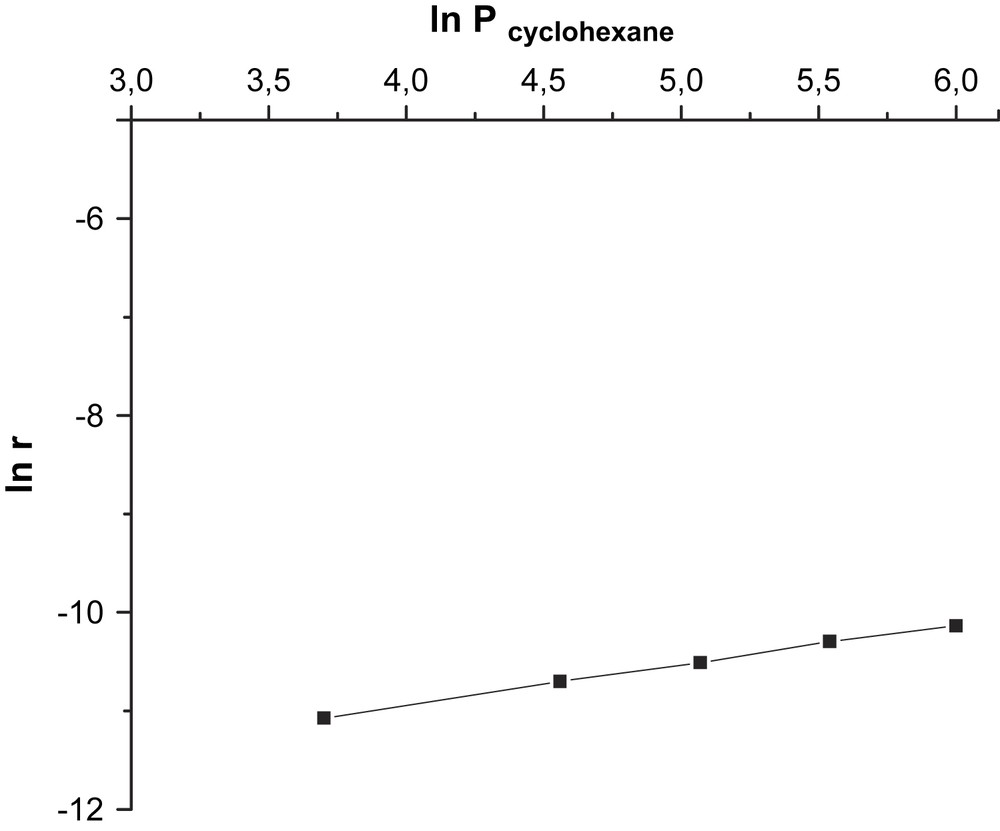
Détermination graphique de l'ordre partiel de la réaction par rapport au cyclohexane.
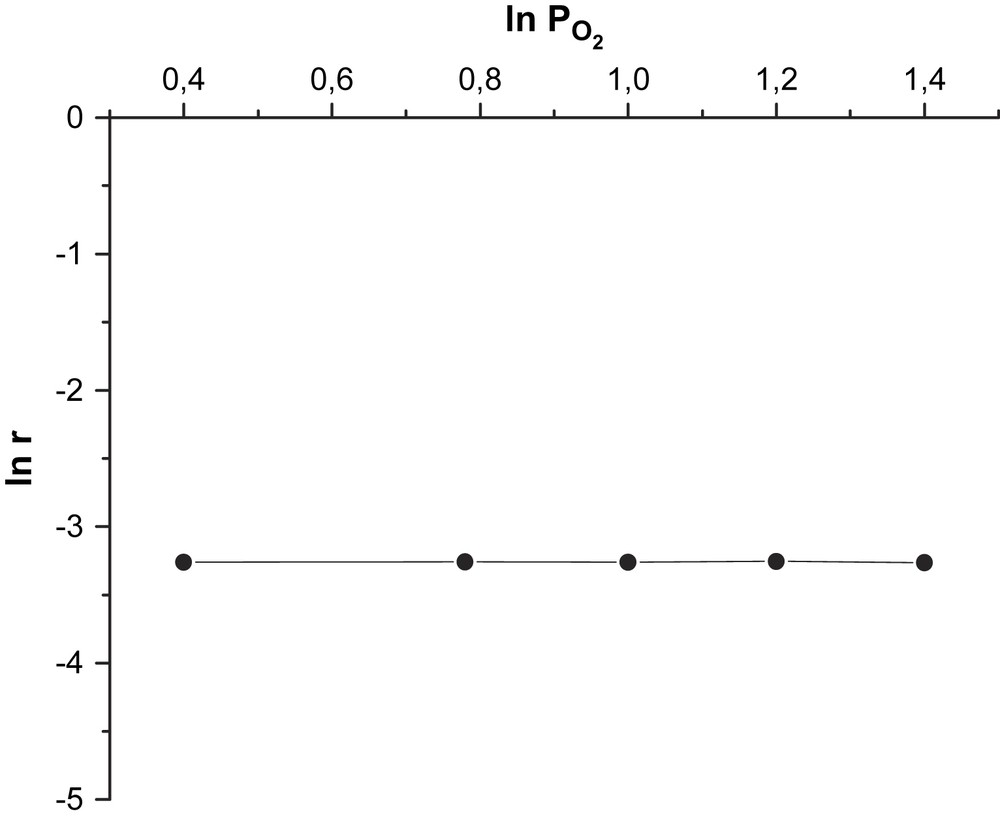
Détermination graphique de l'ordre partiel de la réaction par rapport à l'oxygène.
En régime stationnaire, ces vitesses sont égales. Par ailleurs, le bilan matière relatif aux sites réactionnels permet d’écrire :
| [SO] + [S] = [S]0 |
| (3) |
| (4) |
Si l’étape de réduction est limitante, on peut écrire : k(ox) >> k(réd) PO2, d'où :
où k est la constante de vitesse du processus global. La cinétique est bien du premier ordre par rapport au cyclohexane. Par ailleurs, les valeurs des constantes de vitesse des étapes de réduction et de ré-oxydation (Figs. 1 et 2), déterminées graphiquement en linéarisant l’équation de la vitesse globale :
| K(réd) = 4,3 × 10−4 mol min−1 g−1 torr−1 |
| K(ox) = 0,58 mol min−1 g−1 torr−1 |
Les valeurs relatives des constantes de vitesse montrent bien que l’étape de réduction du catalyseur est l’étape déterminante.
3.1.2 Énergie d'activation apparente
La conversion en cyclohexane au contact des catalyseurs CsFePMo12 et CsFePMo11V est maintenue inférieure à 10% dans un domaine de température compris entre 250 et 350 °C, avec un même temps de contact. Les résultats catalytiques sont rapportés dans le Tableau 1. Les énergies d'activation apparente, déterminées dans les conditions de cinétique initiale avec une pression en cyclohexane considérée comme constante, sont de 12,8 et 14,2 kcal mol−1 pour CsFePMo12 et CsFePMo11V, respectivement. Elles sont du même ordre de grandeur, indiquant ainsi des mécanismes très semblables pour les deux catalyseurs. Elles sont aussi en bon accord avec les données de la littérature (12–23 kcal mol−1) [19,20].
Évolution de la conversion en fonction de la température de la réaction sur CsFePMo12 et CsFePMo11V
| T (°C) | 250 | 275 | 300 | 325 | 350 |
| Conv. (%) (CsFePMo12) | 0,9 | 1,7 | 2,9 | 5,8 | 7,5 |
| Conv. (%) (CsFePMo11V) | 1,0 | 1,7 | 3,3 | 6,0 | 9,3 |
3.1.3 Effet du temps de contact
L'étude de l'influence du temps de contact sur les propriétés catalytiques du solide CsFePMo12 a été réalisée à 350 °C sur une masse de catalyseur de 100 mg, avec un débit des réactifs variant de 0,72 à 2,4 l h−1. Les courbes des sélectivités relatives en fonction du temps de contact (r) sont représentées sur la Fig. 5. Elles montrent que, lorsque le temps de contact tend vers zéro, γ tend vers zéro et 1/γ vers l'infini. Ces résultats impliquent que le benzène et le cyclohexène sont issus du cyclohexane par des réactions successives (Schéma 1) et non des réactions parallèles (Schéma 2). Dans ce dernier cas, la sélectivité devrait tendre vers une valeur finie différente de zéro lorsque le temps de contact tend vers zéro. Le cyclohexène serait bien le précurseur du benzène lors de l'ODH du cyclohexane sur CsFePMo12. Ce résultat est confirmé par la forte diminution de la sélectivité en cyclohexène avec la conversion, qui est typique d'un processus d'oxydation consécutive (Fig. 6) [6,21,22]. Ces résultats sont en accord avec ceux de Patcas [6], Russel [19] et Lock [23], qui ont observé à l'aide d'un spectromètre de masse, lors de la réaction de déshydrogénation du cyclohexane sur le platine, la formation de cyclohexène et de cyclohexadiène. Pour ces auteurs, le benzène n'est pas issu directement du cyclohexane, mais, par une suite de réactions successives, de la transformation du cyclohexane en cyclohexène, puis en cyclohexadiène, et enfin en benzène. Notons que, selon V. Haensel [24], la formation du benzène s'effectuerait en une seule étape réactionnelle (C6H12 → C6H6), qui se justifierait par l'absence du cyclohexène et du cyclohexadiène dans le mélange réactionnel. Toutefois, en accord avec Smith et al. [25], l'absence d'intermédiaires dans le mélange réactionnel ne prouve pas que le mécanisme successif soit impossible.
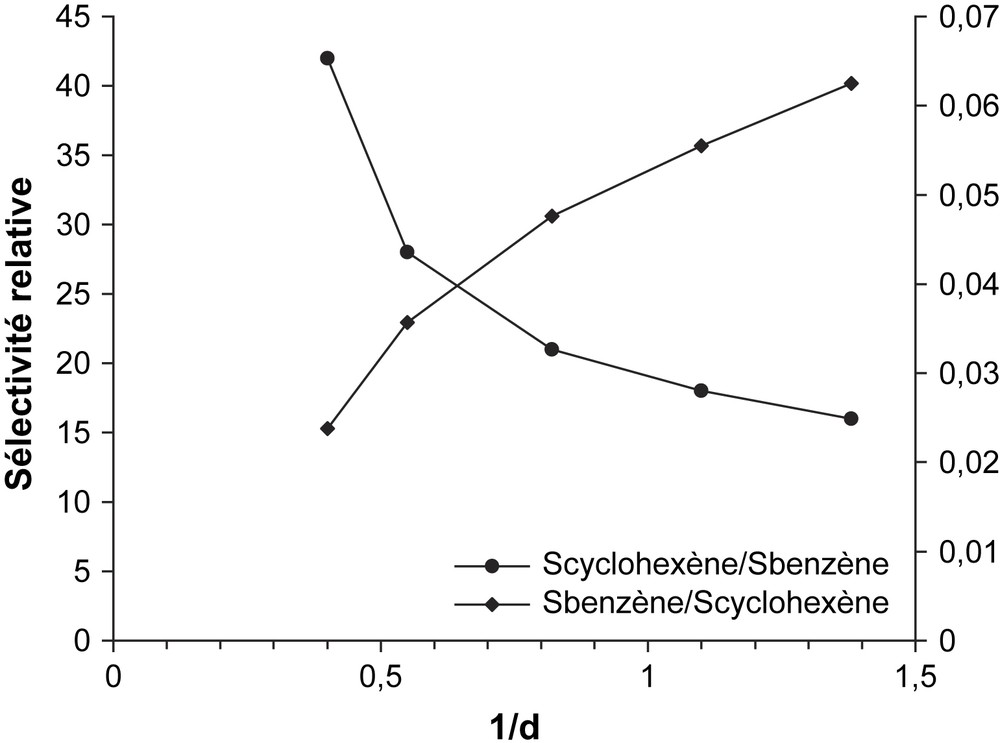
Sélectivités relatives en fonction du temps de contact (τ).

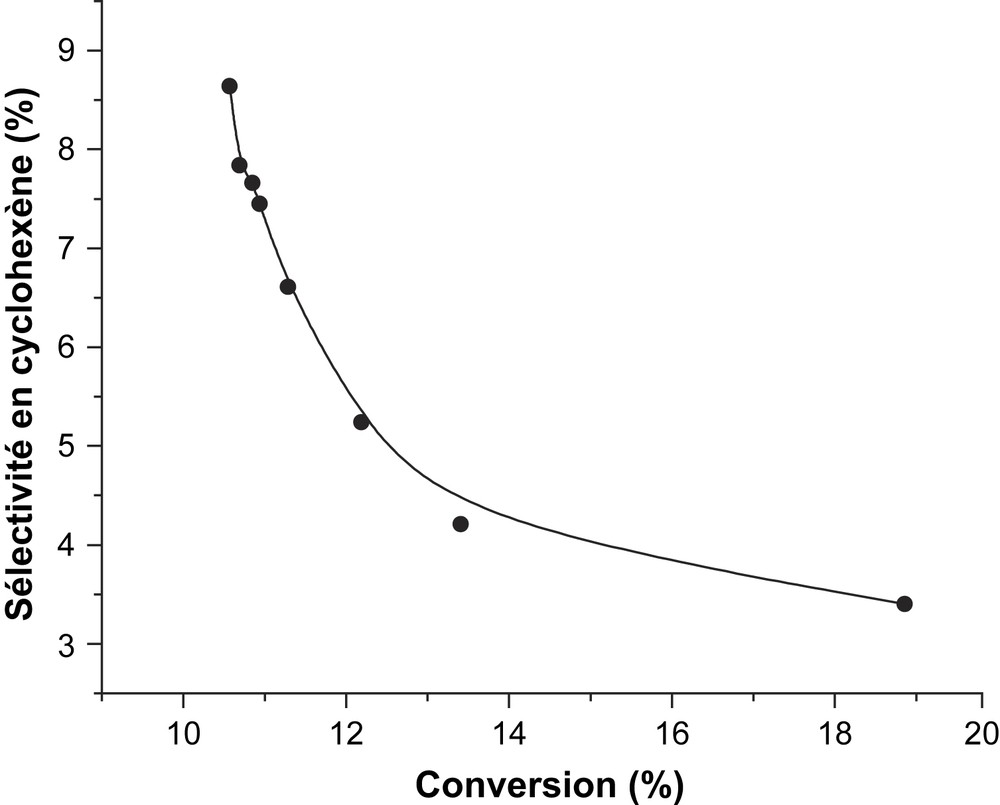
Évolution de la sélectivité en cyclohexène en fonction de la conversion à 350 °C sur CsFEPMo12.
3.2 Étude mécanistique
Les tests catalytiques ont été réalisés sur CsFePMo12 à 350 °C, avec un temps de contact de 0,5 h l−1. Dans le but de préciser les mécanismes réactionnels mis en jeu, nous avons examiné la réactivité des produits formés au cours de la réaction, comme le benzène, le cyclohexène ou le cyclohexadiène. Ce dernier produit est probablement un intermédiaire issu du cyclohexène et précurseur du benzène. La réactivité de produits oxygénés tels que le cyclohexanol et la cyclohexanone a été également examinée. La formation de cyclohexanone a été signalée dans l'ODH du cyclohexane [26]. Les tests de réactivité du benzène et de la cyclohexanone ont montré que ces deux réactifs sont stables dans nos conditions opératoires, ce qui suggère que le benzène est bien le produit final de la réaction de l'ODH du cyclohexane et que la cyclohexanone n'est pas un intermédiaire réactionnel.
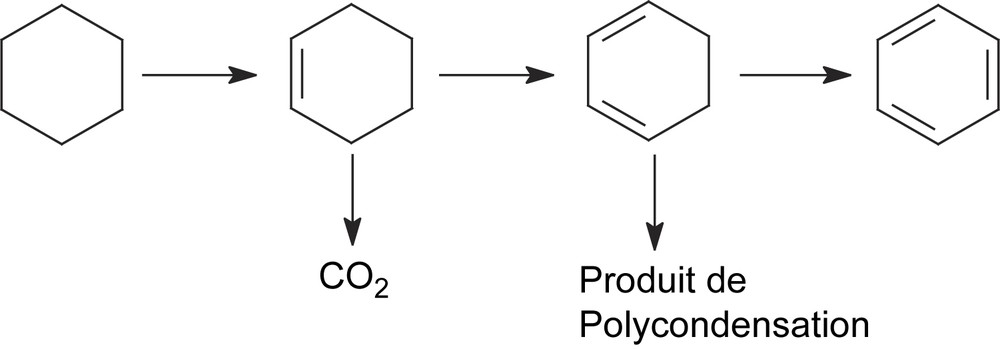
L'oxydéshydrogénation du cyclohexène sur CsFePMo12 à 350 °C a conduit à la formation de benzène, avec une sélectivité de 90% et à celle de CO2 (environ 6% de sélectivité). Le cyclohexadiène se trouve à l’état de trace (Tableau 2, Fig. 7). Aucune trace du produit de polymérisation, de CH4 ou de n-hexane n'est détectée dans le mélange réactionnel. L'obtention quantitative de benzène à partir de cyclohexène confirme que ce dernier est le précurseur du premier, ce qui est en bon accord avec notre étude cinétique ci-dessus. La détection de traces de cyclohexadiène, plus réactif que le cyclohexène, suggère qu'il pourrait aussi être un intermédiaire dans la formation du benzène. Le dioxyde de carbone CO2 serait, lui aussi, issu de l'oxydation du cyclohexène. Par ailleurs, l'allure des courbes de sélectivité au cours du temps (Fig. 7) semble indiquer que le benzène et le CO2 sont formés par des voies parallèles. L'absence de n-hexane dans le mélange réactionnel était attendue, car il ne peut provenir que du cyclohexane, lui-même absent dans le mélange gazeux introduit. La même explication est valable pour l'absence de formation de méthane, qui serait produit principalement par craquage ou combustion du cyclohexane. Ces résultats catalytiques conduisent à proposer le schéma réactionnel suivant (Schéma 3) :
Réaction du cyclohexène sur CsFePMo12, Tp = Tr = 350 °C, N2/O2 = 1, d = 2 l h−1, Ts = 7 °C
| T (min) | 30 | 60 | 90 | 120 | 150 | 180 | 210 | 240 |
| Conversion (%) | 94,4 | 96,1 | 97,1 | 98,1 | 98,7 | 98,7 | 99,3 | 99,4 |
| Sel. cyclohexadiène (%) | 0,8 | 0,1 | 0,3 | 0,4 | 0,4 | 0,5 | 0,6 | 0,7 |
| Sel. benzène (%) | 90,0 | 90,1 | 89,9 | 89,8 | 89,8 | 89,7 | 89,6 | 89,4 |
| Sel. CO2 (%) | 9,2 | 9,9 | 9,8 | 9,8 | 9,9 | 9,9 | 9,8 | 9,8 |
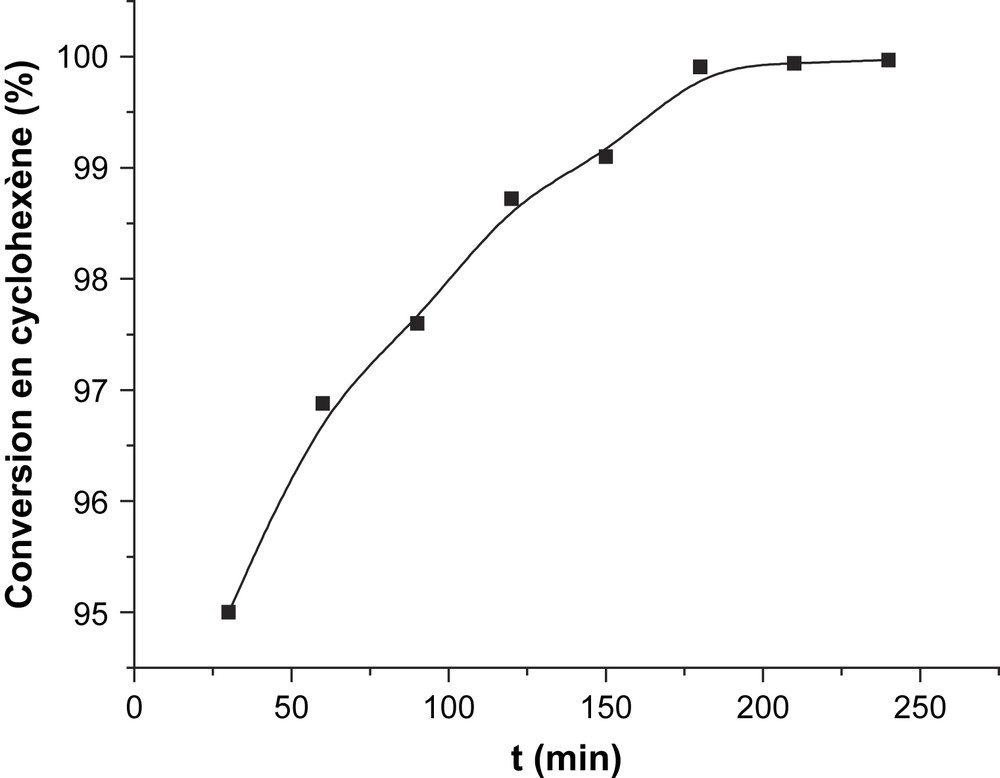
Évolution de la conversion du cyclohexène en fonction du temps sur CsFePMo12 ; Tp = Tr = 350 °C, N2/O2 = 1, d = 2 l h−1, Ts = 7 °C.
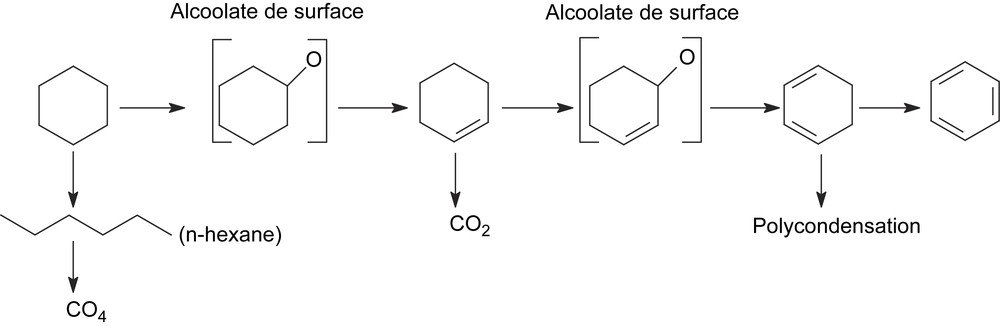
La décomposition du cyclohexanol à 350 °C sur CsFePMo12 conduit à la formation de cyclohexène, de benzène et de cyclohexanone. La formation de ces produits semble indiquer que le cyclohexanol, ou l'espèce adsorbée correspondante, serait un intermédiaire de l'ODH du cyclohexane. Il est d'ailleurs plus réactif que le cyclohexane. En effet, dans les mêmes conditions opératoires (350 °C), il est converti à 20%, alors que le cyclohexane n'est transformé qu’à un taux de 10%. Sa très forte réactivité pourrait expliquer qu'il n'apparaisse pas dans le mélange réactionnel. Il resterait adsorbé sous forme d'alcoolate à la surface. Le mécanisme de formation du cyclohexène serait donc le suivant :
| C6H11OH → [C6H11O] → C6H10 |
La déshydratation du cyclohexanol s'effectuerait, comme décrit ci-dessus, sur un site acido-basique par l'intermédiaire de l'alcoolate de surface correspondant :
| C6H11OH + MOsurface → C6H10OMsurface + OHsurface |
| C6H11OMsurface + HOsurface → C6H10 + H2O + MO |
Par analogie, les précurseurs immédiats de la formation du benzène (et du cyclohexadiène aussi) seraient les alcoolates correspondants :
| C6H10 → [C6H9O] → C6H8 → [C6H7O] → C6H6 |
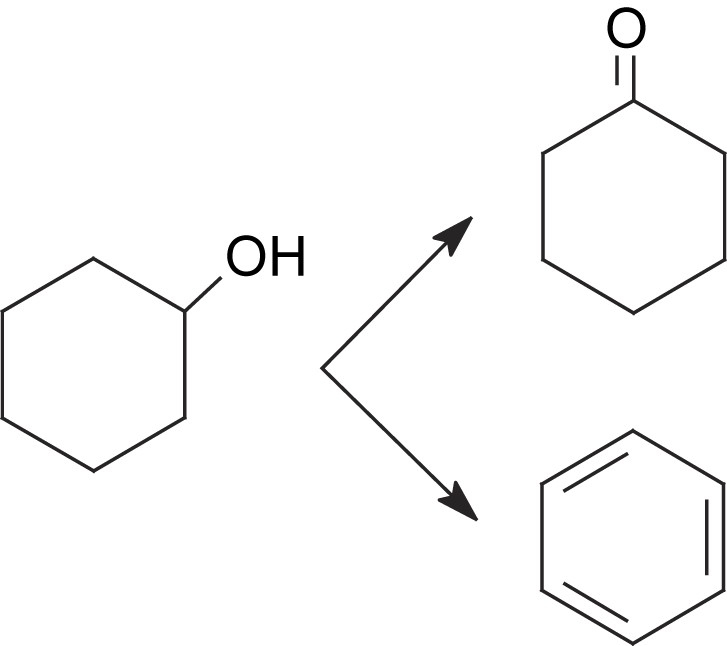
La cyclohexanone semble se former directement à partir du cyclohexanol par un chemin parallèle, car sa sélectivité augmente au cours du temps avec celle du benzène.
La formation de la cyclohexanone est une réaction de déshydrogénation s'effectuant probablement sur un site bifonctionnel basique/oxydant via l'alcoolate correspondant :
| C6H11OH + MOsurface → C6H10OMsurface + OHsurface |
| C6H11OMsurface → C6H10O + MHsurface |
| MHsurface + ½ O2 + HOsurface → H2O + MOsurface |
La conversion du cyclohexanol met ainsi en évidence la compétition de formation du cyclohexène/benzène et de la cyclohexanone sur des sites acido-basiques et/ou oxydants. La réaction est très largement en faveur du cyclohexène et du benzène, ce qui est une indication du caractère très acide du catalyseur CsFePMo12. Ce résultat est en bon accord avec l'absence de détection de cyclohexanone lors de l'ODH du cyclohexane.
Le schéma ci-dessous résume les principales conclusions mécanistiques relatives à l'ODH du cyclohexane sur CsFePMo12 :
4 Conclusion
Les études cinétique et mécanistique ont montré que la réaction d'oxydéshydrogénation du cyclohexane est d'ordre 1 en cyclohexane et d'ordre en oxygène. Cela implique que l’étape déterminante est la réduction de la surface du catalyseur selon un mécanisme de type Mars et Van Krevelen. La réaction produit quantitativement du benzène par des réactions successives impliquant le cyclohexène et le cyclohexadiène comme produits intermédiaires. Les sites actifs seraient bifonctionnels, acido-basiques et oxydants. Les alcoolates de surface seraient les précurseurs des produits insaturés formés.