

1 Introduction
Solar ultraviolet radiation is known to induce reactions between adjacent pyrimidines in DNA, resulting in dimers which can block replication and transcription and are thus potentially lethal and mutagenic [1]. These photoproducts in general are cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6–4) pyrimidone photoproducts, except in the bacterial spores, a dormant form produced by some bacteria such as the Bacillus and Clostridium species [2]. In that case the only photoproduct produced upon exposure to UV light corresponds to two thymines linked by the methyl group of one of the bases [3,4]. The formation of this specific lesion, 5-thyminyl-5,6-dihydrothymine (spore photoproduct, SP) is explained by specific features of the spores, including DNA conformation (A-like form), dehydration, presence of dipicolinic acid in the core and binding of small acid-soluble proteins to DNA [5–8]. In spite of the readily formation of this dimeric lesion, these bacterial spores are extremely resistant to UV radiation since they activate, early in the germination cycle, a very efficient specific repair enzyme, the spore photoproduct lyase (SPL) that directly reverts SP to two unmodified thymines [9,10].
Our current knowledge of the structure and reactivity of the SPL was almost exclusively derived from studies using the enzyme from Bacillus subtilis [11–16]. It was unambiguously shown to be a member of the large “radical-SAM” (S-adenosylmethionine) superfamily of enzymes [17] involved in a variety of biosynthetic pathways and metabolic reactions that proceed via radical mechanisms [18–20]. It contains a catalytically essential [4Fe–4S]2+/+ cluster chelated by the CysXXXCysXXCys sequence, a signature for this class of enzymes, and the reaction is absolutely dependent on S-adenosylmethionine (SAM or AdoMet) [13,14]. Elegant studies, using labelled compounds, from J. Broderick's laboratory have shown that the repair mechanism is likely to involve a 5′-deoxyadenosyl radical (Ado) generated through reductive cleavage of SAM and is initiated by direct C-6 hydrogen atom abstraction by Ado, with the resulting substrate radical undergoing scission to re-generate the two initial thymines (Fig. 1) [14,21]. SAM is suggested to be used by this enzyme as a catalytic cofactor. Our investigations demonstrated that the enzyme is active on SP-containing DNA as well as on a dinucleoside monophosphate SP substrate, suggesting that the active site of the enzyme is designed to flip the thymine dimer to be repaired out of the duplex DNA target and bind it specifically [22].
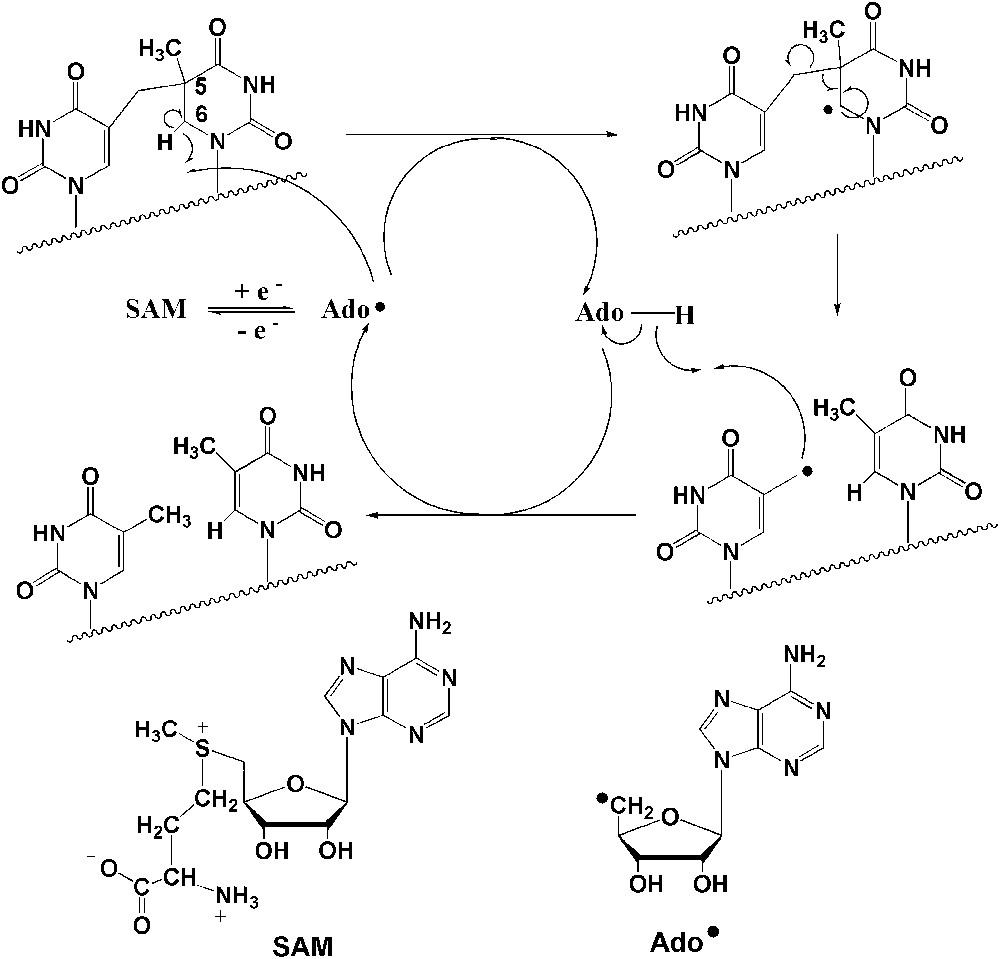
Proposed chemical mechanism for SPL-mediated DNA repair [14].
Unfortunately the SPL from B. subtilis is very difficult to characterize, since only limited amounts are accessible, the protein is very prone to precipitate under various conditions and the iron–sulfur cluster is particularly labile. As a consequence, a number of previous studies were based on poorly active preparations, with low cluster content. In order to obtain protein preparations with sufficient stability, in large amounts, in particular for mechanistic and X-ray crystallography studies, we searched for another source of SPL. We found that the recombinant SPL enzyme from Clostridium acetobutylicum, isolated either aerobically or anaerobically from overexpressing E. coli, was filling these criteria. We report here a complete spectroscopic and chemical characterization of this enzyme. In particular we show for the first time that SAM binds to the cluster as observed in the case of other members of the “Radical-SAM” enzyme family such as the activases of pyruvate formate lyase and ribonucleotide reductase [23–26].
2 Experimental procedures
2.1 Materials
Strains: E. coli DH5α was used for routine DNA manipulations. E. coli C41 (DE3) was used for SPL overexpression. Enzymes, oligonucleotides, and culture media were purchased from Invitrogen (Cergy-Pontoise, France). T4 DNA ligase was from Promega, Inc. Bacterial alkaline phosphatase was from Amersham-Pharmacia, Inc. and plasmid DNA purification kit, Qiaprep™, was from Qiagen. DNA fragments were extracted from agarose gel and purified with High Pure PCR Product Purification Kit (Roche, Inc.). DNA sequencing was performed by Genome Express (Grenoble, France). TpT was prepared as described in [27].
2.2 Cloning and construction of SPL-overexpressing plasmids
The SplB gene, encoding SPL, was amplified by a polymerase chain reaction (PCR)-based method using C. acetobutylicum ATCC824 genomic DNA as a template. The following primers were used: 5′- ggtaatccatATGgaaaatatgtttagaagagttatatttgaaaagaaag -3′ (NdeI site underlined, ATG codon in upper case) hybridized to the non-coding strand at the 5′ terminus of the gene and 5′-gccggatccttaaattatatacttaattgttgccttgttaaaatataaatttatattg-3′ (BamHI site underlined) hybridized to the coding strand. PCR was run on a Stratagene RoboCycler Gradient 40 machine. The PCR product was digested with BamHI and NdeI and then ligated with T4 DNA ligase into the pET28a plasmid (containing the hexahistidine tag on its N-terminal side), which had been previously digested with the same restriction enzymes. The cloned gene was entirely sequenced to ensure that no error was introduced during PCR reaction. The plasmid was then named pET-SPLCA.
2.3 Protein expression
E. coli C41 (DE3) was transformed by pET-SPLCA and then grown overnight at 37 °C in LB medium (100 mL) supplemented with kanamycin (50 μg/mL). The overnight culture was used to inoculate fresh LB medium (10 L) supplemented with the same antibiotic, bacterial growth was allowed to proceed at 37 °C until OD600 reached 0.9. To reduce formation of inclusion bodies, protein expression was performed at 18 °C and induced by adding 500 μM of IPTG. Cells were collected after 18 h of culture by centrifugation at 4000 × g at 4 °C for 30 min and resuspended in buffer A (50 mM Tris–HCl, pH 7.5, 200 mM KCl) containing 0.5% Tween 20, 0.5% Triton x100 and anti-proteases cocktail Complete EDTA-free (Roche). The cells were disrupted by sonication and centrifuged at 220,000 × g at 4 °C for 1 h 30 min. The solution obtained was then loaded onto a Ni-NTA Superflow column (Qiagen) which had been previously equilibrated with buffer A. The column was washed extensively with the same buffer. Non-specifically adsorbed proteins were eluted by a wash step with buffer A containing 30 mM imidazole and then the SPL was eluted with a gradient of buffer A reaching 0.5 M imidazole. Fractions containing SPL were pooled, filtrated and loaded immediately on a Hi Prep desalting column (GE Healthcare) to remove imidazole and prevent from precipitation. The fractions containing SPL were concentrated in an Amicon cell fitted with a YM30 (Spectrapor) membrane and 3 mM DTT were added before freezing.
For anaerobic purification of the SPL, the same protocol was applied until the resuspension of cells, except that the growth medium was supplemented with 100 μM Fe citrate or 35 μM 57FeCl3 for Mössbauer spectroscopy experiments. Bacterial growth and induction were performed as for the aerobic procedure. Cells were resuspended in the anaerobic glove box with buffer A and anti-proteases cocktail Complete EDTA-free (Roche). The following steps were carried out in the glove box. The cells were incubated under agitation with lysozyme (1 mg/mL) for 30 min and Triton x100 was added at a final concentration of 0.5%. The mixture was incubated for one more hour, then transferred into ultracentrifugation tubes and subjected to three cycles of freezing/thawing before centrifugation as stated above. The solution obtained was loaded anaerobically on a Ni-NTA Superflow column (Qiagen) equilibrated with buffer A and purified as described above. The fractions containing SPL were desalted on NAP 25 to remove imidazole, concentrated on Microcon YM 30 (Amicon) and 3 mM DTT were added. The preparation could be used for activity assays and Mössbauer spectroscopy.
2.4 Iron and sulfide binding to SPL
The procedure was carried out anaerobically in a glove box (Jacomex B553 (NMT)) as previously described for SPL from B. subtilis [22].
2.5 Production of SPTpT substrates
The spore photoproduct of the dinucleoside monophosphate thymidylyl-(3′-5′)-thymidine was prepared as previously described [22].
2.6 SPL activity
To assay SPL activity, we followed the same procedure as for B. subtilis SPL [22], using DTT, dithionite and SAM in the same proportions. At each time point (0, 30, 60 and 120 min), 20 μL of the solution were transferred to an Eppendorf tube and the reaction stopped by flash-freezing in liquid nitrogen. The same conditions were used to assay substrate affinity using different concentrations of SPTpT.
2.7 HPLC-mass spectrometry analysis
Conversion of the spore photoproduct (SPTpT) into the unmodified dinucleoside monophosphate (TpT) in SPL-treated samples was quantified by HPLC coupled to tandem mass spectrometry (HPLC-MS/MS), using the same conditions as for samples treated by SPL from B. subtilis [22].
2.8 Protein analysis
Protein concentration (by monomer) was determined by the method of Bradford [28]. Protein-bound iron [29] and labile sulfide [30] concentrations were determined according to standard procedures.
2.9 UV–visible absorption spectroscopy
UV–visible absorption spectra were recorded with a Cary 1 Bio (Varian) spectrophotometer.
2.10 Electron paramagnetic resonance (EPR)
Reconstituted SPL (200 μM, 3.9 iron/protein) or SPL purified anaerobically (375 μM, 1.6 iron/protein) were reduced with 3 mM dithionite under anaerobic conditions for 20 min and frozen inside the glove box. X-band EPR spectra were recorded on a Brucker Instruments ESP 300D spectrometer equipped with an Oxford Instruments ESR 900 flow cryostat (4.2–300 K). Spectra were quantified under non-saturating conditions by double integration against a 1 mM Cu-EDTA standard.
2.11 Mössbauer spectroscopy
For Mössbauer measurements, reconstituted SPL (4.56 mg, 4.01 iron/protein) or anaerobic SPL (3.3 mg, 1.6 iron/protein) was prepared as described above in a final volume of 400 μL. The protein solution was transferred into a Mössbauer cup and frozen in liquid nitrogen. 57Fe-Mössbauer spectra were recorded at zero magnetic field on a spectrometer operating in constant acceleration mode using an Oxford cryostat that allowed temperatures from 1.5 to 300 K and a 57Co source in rhodium. Isomer shifts are reported relative to metallic iron at room temperature.
2.12 HYSCORE spectroscopy
For HYSCORE measurements, reconstituted SPL (3.84 mg, 3.9 iron/protein) was prepared as described above in a final volume of 400 μL. Dithionite (2 mM) was added and the protein incubated for 20 min. The mixture was divided into two: the first EPR tube was directly frozen and in the second, 5 mM SAM were added before freezing.
HYSCORE experiments were performed on a Bruker Elexsys E-580 X-band pulsed spectrometer with a Bruker ER4118X dielectric resonator and a continuous flow He cryostat (Oxford Instrument CF935) controlled by an Oxford Instrument temperature controller ITC 503. Experiments were performed at 10 K using the standard four-pulse sequence (π/2-τ-π/2-t1-π-t2-π/2-echo) with a nominal pulse width of 16 ns for π/2 and π pulses, a τ value of 128 ns and a pulse repetition rate of 2 kHz. Unwanted echoes were removed by four-step phase cycling. A 64×64 dataset was recorded with times t1 and t2 incremented in 24 ns steps from an initial value of 200 ns. This dataset was processed using Xepr software (Brucker). The background decay in both dimensions was subtracted using a linear fit followed by apodization with a Hamming window and zero-filling to 512 points in each dimension. The 2D Fourier Transform magnitude spectrum was calculated and presented as a contour plot.
3 Results
3.1 Cloning, overexpression, and purification of recombinant SPL from C. acetobutylicum
The pET-SPLCA plasmid, obtained as described in Section 2, was used to transform E. coli C41 (DE3) strain for production of a protein with a His-tag at the N-terminus part. The introduction of a His tag at the N-terminus has been reported in the case of B. subtilis SPL to exhibit no effect on enzymatic activity [11]. Unlike SPL from B. subtilis, SPL from C. acetobutylicum was mainly recovered in soluble extracts and very little in inclusion bodies. The cell-free extracts were loaded onto an affinity chromatography column (Ni-NTA Superflow – Qiagen) and after an extensive washing with buffer containing 30 mM imidazole, the protein eluted at 300 mM imidazole. In order to avoid precipitation of the Ni-NTA eluted protein due to the presence of imidazole, a desalting purification step was immediately applied to the solution. About 50 mg of SPL could be obtained from 10 L of culture. The purity of the purified protein was judged by SDS electrophoresis analysis to be around 95% and its apparent molecular weight was 41 kDa, as expected from the amino-acid sequence (Fig. 2-I). The purified protein was slightly brown and was found to contain substoichiometric amounts of iron and sulfur atoms (<0.2 of each atom per equivalent protein). This low amount was possibly due to the loss of the oxygen-sensitive cluster during aerobic purification, as often observed with [Fe–S] proteins, and proved that the as-isolated protein was mainly under the apo form. As a matter of fact, when the extraction and purification were carried out in an anaerobic glove box, the purified SPL protein was much more colored and was found to contain up to about 2 Fe and S atoms per monomer. This preparation will be named anaerobic SPL in the following.
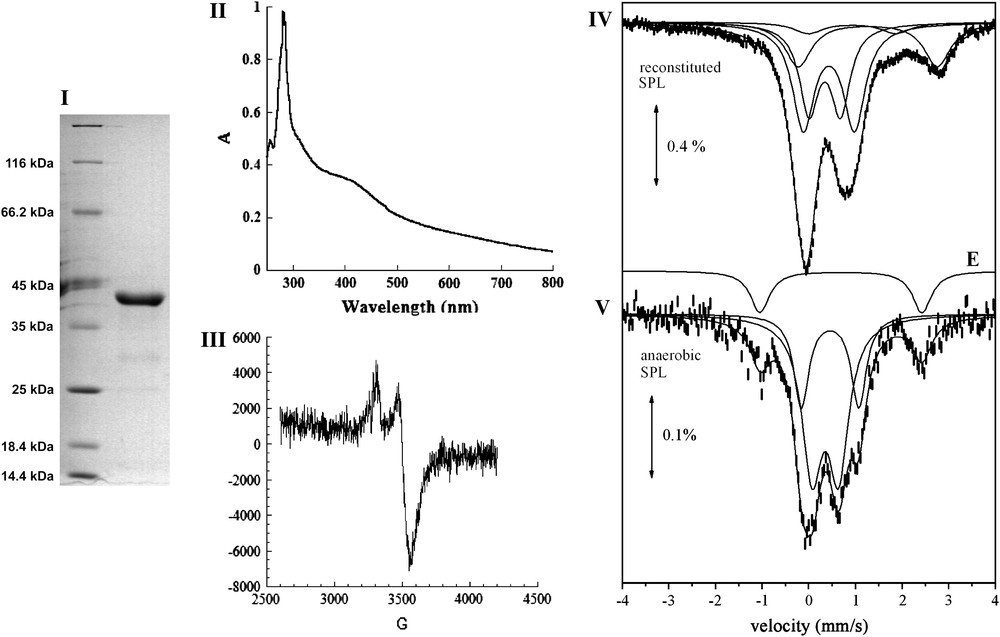
Biochemical and spectroscopic characterization of SPL. (I) 12% SDS-PAGE: Lane 1: Molecular Weight markers, Lane 2: Reconstitued SPL. (II) UV–vis absorption spectrum of the anaerobically reconstituted SPL protein from Clostridium acetobutylicum (SPL 0.9 mg/mL in 100 mM Tris–HCl, pH 8, 50 mM KCl); (III) X-band EPR spectrum of the anaerobically reduced reconstituted SPL (200 μM in 100 mM Tris–HCl, pH 8, 50 mM KCl). The spectrum was recorded under the following conditions: temperature: 10 K; microwave power: 0.2 mW; modulation amplitude: 10 G; (IV) and (V), 57Fe-Mössbauer spectra at 4.2 K of 277 μM reconstituted SPL (IV) and 208 μM anaerobic SPL (V). Solid lines represent theoretical simulations assuming different species (see text) with the parameters quoted in Table 1. Doublet E is displayed above the spectrum of the anaerobic SPL.
3.2 Reconstitution and characterization of the iron–sulfur center of SPL
The aerobically obtained apo form was treated with iron and sulfide salts under anaerobic conditions to convert it into the holoform using previously described procedures [31]. After reconstitution of the cluster, the enzyme was found to contain approximately 3.9 iron and sulfur atoms per protein monomer. In the following, this preparation will be named reconstituted SPL.
The UV–vis light absorption spectrum of the reconstituted protein, shown in Fig. 2-II, is very similar to spectra of other [Fe–S] proteins of the “Radical-SAM” enzyme superfamily (RNR activase, MiaB, HemN) [31–33] with a significant absorption band at 420 nm. It is thus consistent with the presence of a large proportion of polypeptides carrying a [4Fe–4S] cluster but also possibly a [2Fe–2S]. The UV–visible spectrum of the anaerobic SPL preparation was similar to that shown in Fig. 2-II. Exposure of the protein to air resulted in a rapid degradation of the cluster, as measured by the decrease of the absorption band at 420 nm (data not shown). During anaerobic reduction of the reconstituted protein with an excess of sodium dithionite, the solution bleached, with a concomitant decrease of the absorption band at 420 nm. The resulting reduced solution displayed an axial EPR spectrum with g values at g = 2.03(7) and 1.93(7) (Fig. 2-III). The temperature dependence and microwave power saturation properties of the EPR signals were characteristic of the S = 1/2 ground state of a [4Fe–4S]+ cluster (data not shown). Integration of the signal indicated that only 10% of total iron present in the sample was in the form of a reduced [4Fe–4S]+ cluster. This likely results from incomplete reduction of the cluster due to a low redox potential, as generally observed in “Radical-SAM” enzymes, and partly also from partial degradation of the cluster under reducing conditions.
The [Fe–S] clusters were further analyzed by Mössbauer spectroscopy using a reconstituted protein sample (277 μM, 4.01 iron/monomer) that had been obtained by incubating anaerobically the apo form with 57Fe and sulfide for 15 h at 4 °C. In Fig. 2-IV the spectrum recorded at 4.2 K (upper spectrum) is shown. An almost identical spectrum was obtained at 78 K. The spectrum is remarkably similar to the one observed for SPL from B. subtilis reconstituted with Fe and S in a similar way [22]. As in the previous case the spectrum indicates the presence of at least four different species giving rise to quadruple doublets which significantly overlap. By keeping the same assignment as before [22], doublet A is attributed to a [4Fe–4S]2+ (S = 0) cluster and doublet B to a [2Fe–2S]2+ (S = 0) cluster. We recall that the large degree of overlapping between doublets A and B does not allow us to make an accurate determination of their relative ratio. We estimate that the [4Fe–4S]2+ clusters account for 42% of total iron and the [2Fe–2S]2+ clusters for 31%. Doublet D (21% of total iron) is attributed to octahedral high spin ferrous impurities. Doublet C accounts for minor species (6% of total iron) and has parameters which can be attributed to a high-spin ferrous species as well.
The 4.2 K spectrum from an anaerobic SPL enzyme sample is also shown in Fig. 2-V. A similar spectrum was obtained at 78 K. The low iron content in this sample (about 335 μM) leads to a relatively noisy spectrum. However it is clear that the majority of the iron (85%) gives rise to a spectrum which can be considered as the superposition of two doublets similar to A and B of the reconstituted sample. Therefore we conclude that the spectrum contains [4Fe–4S]2+(S = 0) (27% of total iron) and [2Fe–2S]2+(S = 0) clusters (58% of total iron). Apparently, the amount of [2Fe–2S]2+ clusters is larger in the anaerobic SPL sample. The rest of the iron (15%) gives rise to a doublet (E) with parameters which are typical for high-spin ferrous species in a tetrahedral environment with S donors.
As a conclusion, the Mössbauer spectra from reconstituted SPL preparations indicate the presence of [2Fe–2S]2+ and [4Fe–4S]2+ clusters. Anaerobic SPL enzyme containing 57Fe also contained the two types of clusters, as shown by Mössbauer spectroscopy, with [2Fe–2S]2+ ones in larger proportions (Table 1).
Parameters and relative amounts of Mössbauer doublets obtained by simulations for reconstituted and anaerobic SPL from C. acetobutylicum
| Sites | Species | Reconstituted SPL | Anaerobic SPL | ||||
| δ (mm/s) | ΔEQ (mm/s) | Area (%) | δ (mm/s) | ΔEQ (mm/s) | Area (%) | ||
| A | [4Fe–4S]2+ | 0.43(2) | 1.09(10) | 42(5) | 0.45(3) | 1.22(10) | 27(5) |
| B | [2Fe–2S]2+ | 0.34(2) | 0.67(5) | 31(5) | 0.35(3) | 0.56(5) | 58(5) |
| C | X | 0.92 | 1.97 | 6(2) | – | ||
| D | Octahedral Fe2+(S = 2) | 1.26 | 2.89 | 21(2) | – | ||
| E | Tetrahedral Fe2+(S = 2) | – | 0.68(2) | 3.47(3) | 15(2) |
3.3 Characterization of SAM–[4Fe–4S]+ complex by HYSCORE spectroscopy
It is now generally accepted that in “Radical-SAM” enzymes, a [Fe–S]–SAM complex involving coordination of SAM to the unique Fe site through its aminocarboxylate moiety is formed as a reaction intermediate [18]. This complex has been directly characterized by spectroscopic methods in the case of the activase of pyruvate formate lyase and of lysine aminomutase [25,26,34] and by X-ray crystallography in the case of biotin synthase, MoaA enzyme and coproporphyrinogen oxidase for example [35–37]. In order to check whether such a [Fe–S]–SAM complex is also generated in the SPL enzyme, we used HYSCORE spectroscopy, a two-dimensional pulsed EPR technique previously used to investigate another “Radical-SAM” enzyme, the activase of ribonucleotide reductase [23]. One of the main advantages of HYSCORE is its ability to sort three types of nuclei: the strongly and weakly coupled ones and the “distant” nuclei, which are characterized by very low hyperfine couplings. In the latter case, the corresponding peaks lie on the diagonal of the (+, +) quadrant, whereas the strongly and weakly coupled nuclei appear in the (−, +) and the (+, +) quandrants, respectively [38,39].
The HYSCORE spectrum of the anaerobically reduced SPL presented no cross-correlation peaks in the (−, +) quadrant. In the (+, +) quadrant, only peaks from distant 13C carbon atoms present in natural abundance and from 1H hydrogen atoms are observed (Fig. 3A). Upon anaerobic addition of SAM to a solution of reconstituted SPL in the reduced S = 1/2 form, the HYSCORE spectrum was drastically modified (Fig. 3B) and a symmetrical set of new features appeared in the (−, +) quadrant. These peak patterns and their position are characteristic of a strongly coupled nitrogen atom (an I = 1 nucleus with quadrupolar coupling). From the position of the so-called double quanta double quanta correlation peaks (dqdq in Fig. 3B) at (9.5; −5.7) and (5.7; −9.5) MHz, it is possible to obtain a good estimation of the isotropic hyperfine coupling aN using:
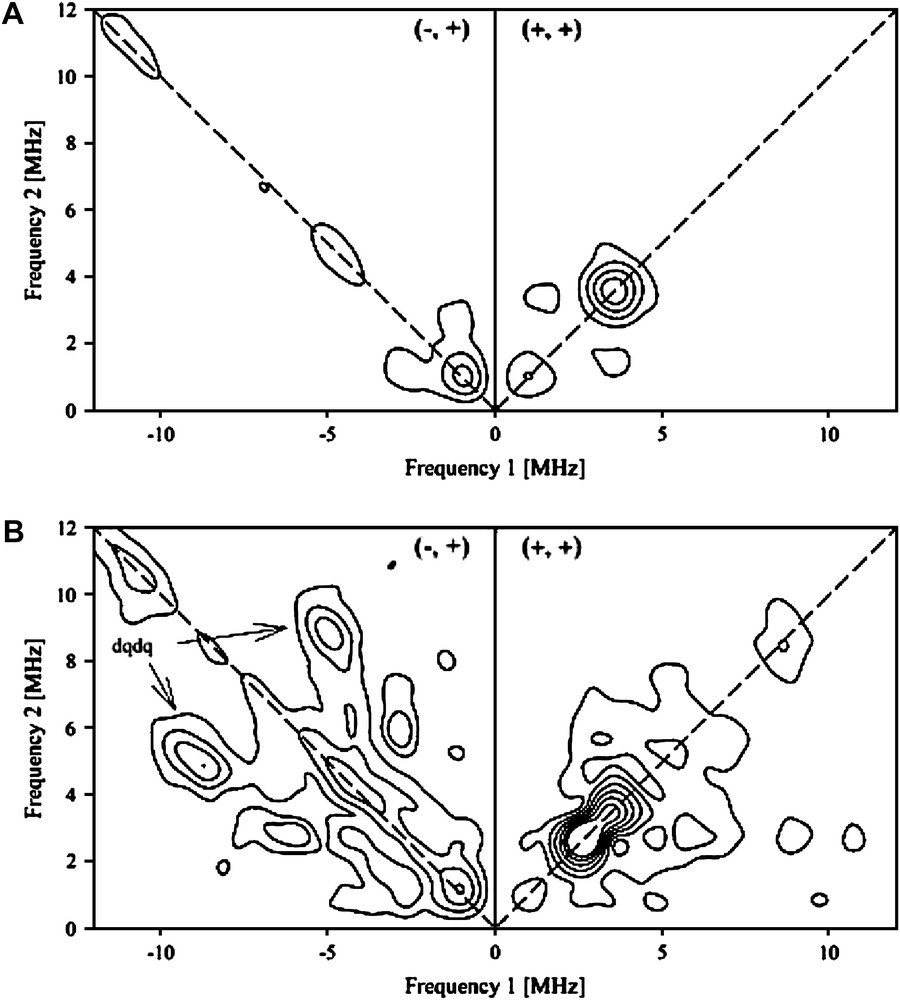
HYSCORE spectra of the anaerobically reduced reconstituted SPL (240 μM) recorded at g = 1.93, before (A) and after (B) addition of 10 molar excess of SAM.
The value obtained (|aN| = 6.5 MHz), when scaled by γ15N/γ14N, gives a coupling constant of 9.1 MHz, which is very close to those obtained by HYSCORE spectroscopy for the anaerobic ribonucleotide reductase activating enzyme (|a15N| = 8.9 MHz) [23], and by 15N ENDOR spectroscopy for the lysine aminomutase (|a15N| = 9.1 MHz) [34]. This strongly suggests that the amino group of SAM is coordinated to the unique Fe of the FeS center of SPL in a manner similar to that demonstrated in related enzymes.
3.4 In vitro DNA repair activity of the reconstituted SPL
In order to characterize the catalytic activity of SPL from C. acetobutylicum, we used the modified dinucleoside monophosphate SPTpT in a pure form as a substrate. SPTpT was prepared by UV irradiation of dry films of the dinucleoside monophosphate TpT in the presence of DPA, as already described [22]. The reconstituted SPL (1 μM) was incubated together with 1.5 mM dithionite as a reducing agent, 1.5 mM SAM and 10 μM SPTpT under strict anaerobic conditions. The reaction mixture was then assayed at time intervals by HPLC-MS/MS for both substrate conversion and TpT (repaired dinucleotide) product formation. Indeed, these compounds can be easily separated by HPLC and identified, as they display characteristic well-defined mass spectrometric features. A typical time curve for SPL-dependent conversion of SPTpT to TpT is shown in Fig. 4. The results showed clearly that SPTpT could be totally converted by the enzymatic reaction into the expected repaired TpT product after 60 min reaction with an initial rate of approximately 0.2 mol/mol enzyme/min. Kinetic parameters were determined using increasing concentrations of the substrate. The Km value was 6 μM, identical to that obtained with the B. subtilis enzyme, and the Vm value was 0.6 mol/mol/min. No TpT could be detected when protein or SAM was omitted from the reaction mixture (control).
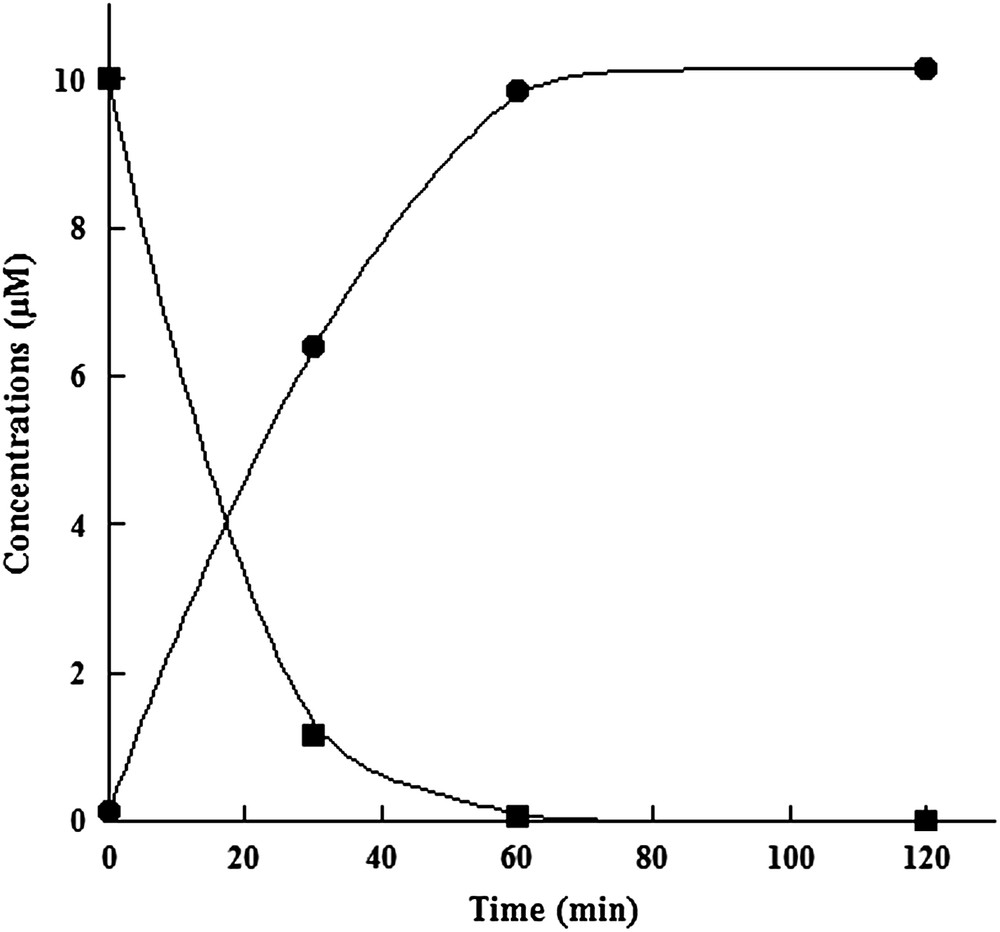
In vitro assay of reconstituted SPL activity with SPTpT as a substrate. Time-dependent enzymatic SPTpT repair (■) and TpT formation (●).
These results demonstrate that SPTpT is a substrate for SPL from C. acetobutylicum and that SPL functions catalytically with such a substrate.
4 Discussion
This work is the result of a search for an abundant source of SPL which would be compatible with spectroscopic, mechanistic and crystallographic studies. Indeed, until very recently, studies of SPL were done on poorly catalytically active and partially cleaved proteins, and proteins with low cluster content. This was due to the fact that the selected enzyme was that from B. subtilis, which is difficult to isolate. Furthermore, it is unstable, with a great tendency to precipitate, and its cluster is highly labile. On the other hand, these initial investigations, mainly by the group of W.L. Nicholson, provided the foundations for investigations of this fascinating DNA-repair enzyme [10,11,13,15]. In 2006, J. Broderick et al. reported a novel preparation for the B. subtilis recombinant enzyme in an active form, allowing the production of 25 mg pure protein from 10 L of growth media [21]. The anaerobic procedure used for the purification explained why the protein contained three Fe and three S atoms per monomer, a significant part (up to 35%) of them being in the form of [3Fe–4S] clusters. In the absence of Mössbauer spectroscopic study of this preparation, it is impossible to evaluate its cluster homogeneity. The addition of SAM to the purified protein was shown to increase its solubility.
Also in 2006, we reported an in vitro study of SP repair using a synthetic dimeric substrate and an SPL preparation from B. subtilis [22]. In order to have full content of Fe and S (about 4 per monomer), we used a reconstituted and active form of SPL, obtained by anaerobic treatment of the apo form by ferrous and sulfide salts. This form was found to be more soluble and stable. This study provided the first characterization of an SPL by Mössbauer spectroscopy, which demonstrated the presence of both [2Fe–2S] and [4Fe–4S] clusters. This is a characteristic of “Radical-SAM” enzymes and is probably a consequence of the lability of the [4Fe–4S] cluster. On the other hand, [2Fe–2S] clusters could be intermediates during [4Fe–4S] clusters' assembly and thus their presence would reflect an incomplete reconstitution.
A last recent study using the SPL from Geobacillus stearothermophilus should be mentioned [41]. However, even though high turnovers are reported, very little information is provided regarding the stability of the protein, the nature and the stability of bound clusters as well as the amount of protein that can be obtained. Furthermore, the protein is a mixture of monomers and dimers together with a population of polypeptides cleaved at the C-Terminus.
We think that the SPL from C. acetobutylicum described here displays significant advantages with respect to the previously studied sources of enzyme.
First, this enzyme is much more soluble and stable. Less precipitation than with B. subtilis enzyme is observed, even in the absence of added SAM. Second, anaerobic purification yields a protein which contains defined clusters, both [2Fe–2S] and [4Fe–4S] with 85% of total iron residing in these clusters. The chemically reconstituted SPL also has about 80% of its Fe in the form of clusters, which is a larger value than in the case of B. subtilis [22]. The presence of a large proportion of [2Fe–2S] clusters in the anaerobically purified SPL was unexpected but might be due to freezing and thawing cycles required for Mössbauer experiments. Third, about 50 mg pure protein can be obtained from 10 L culture, which is the largest yield reported so far.
Furthermore, SPL from C. acetobutylicum displays a slightly greater specific activity as compared to other SPLs and it is interesting to note that it also recognizes and repairs the synthetic SP dimer, previously reported as a defined substrate [22]. This will allow further mechanistic studies.
Finally, HYSCORE spectroscopy has clearly demonstrated, for the first time in SPL, that the cluster of SPL binds the N atom of the aminocarboxylate moiety of SAM. We have also tried to carry out the same study with the B. subtilis, enzyme, but this proved impossible because of the instability of the reduced cluster. This is a nice illustration of the superiority of the C. acetobutylicum enzyme. Since the hyperfine coupling constant aN is identical to that obtained by ENDOR spectroscopy in the case of lysine aminomutase, it is very likely that coordination of SAM to the cluster is the same in the two enzymes. The structure of the complex has been crystallographically determined in the case of lysine aminomutase [34]. This provides an experimental support to the hypothesis that, as in all “Radical-SAM” enzymes, SPL activity begins with binding of SAM to the [4Fe–4S] cluster, followed by electron transfer and reductive cleavage of SAM to generate the 5′-deoxyadenosyl radical, Ado [18–20]. The second step of the reaction which involves radical SP repair by Ado has been quite well characterized by J. Broderick using labelled substrates and cofactors [14,21]. The molecular details of the interactions between the enzyme and the DNA to be repaired await to be identified. The C. acetobutylicum enzyme might be a good choice for this project.