

1 Introduction
Isoprenoids are natural products formally derived from the branched C5-skeleton of isoprene. They include an incredible variety of essential compounds such as carotenoids, cholesterol, hormones, vitamins, the prenyl chains of quinones from electron transport chains (ubiquinones, plastoquinone), of chlorophylls (e.g., phytol) or prenylated proteins, as well as secondary metabolites such as antitumor agents (taxol), rubber, prenyl moiety of meroterpenoids (e.g., indole Catharanthus alkaloids). Isopentenyl diphosphate (IPP, 9) and dimethylallyl diphosphate (DMAPP, 10) represent the biological equivalents of isoprene and serve as the building blocks for all isoprenoids in all organisms. The elucidation of isoprenoid biosynthesis in liver tissue and yeast led to the discovery of the mevalonate pathway in the 1950s [1,2]. The latter pathway was accepted as the universal biosynthetic route for all isoprenoids in all living organisms. A novel metabolic route, the methylerythritol phosphate (MEP) pathway, was, however, discovered in the late 1980s and found in most bacteria [3], green algae [4] and plant chloroplasts [5] as well as in Plasmodium falciparum, the parasite responsible for malaria [6] and questioned the mevalonate paradigm. In this mevalonate-independent MEP pathway, IPP (9) and DMAPP (10) are carbohydrate derivatives. Their synthesis starts from pyruvate (1) and d-glyceraldehyde phosphate (2) via 1-deoxy-d-xylulose 5-phosphate (DXP) (3), 2-C-methyl-d-erythritol 4-phosphate (MEP) (4), 4-diphosphocytidyl-2-C-methyl-d-erythritol (5), 4-diphosphocytidyl-2-C-methyl-d-erythritol 2-phosphate (6), 2-C-methyl-d-erythritol 2,4-cyclodiphosphate (MEcDP) (7) and 4-hydroxy-2-methylbut-2-enyl 1-phosphate (HMBPP) (8) (Fig. 1). The first steps leading from pyruvate 1 and d-glyceraldehyde 3-phosphate 2 to methylerythritol 2,4-cyclodiphosphate 7 are rather well documented (for reviews, see Refs. [7,8]). The last ones, converting the cyclodiphosphate 7 into IPP 9 and DMAPP 10, represented, however, a bottleneck in the full elucidation of the pathway and raised unexpected experimental problems. The approach used to characterize GcpE and LytB, the last two enzymes of the MEP pathway, is described in this contribution.
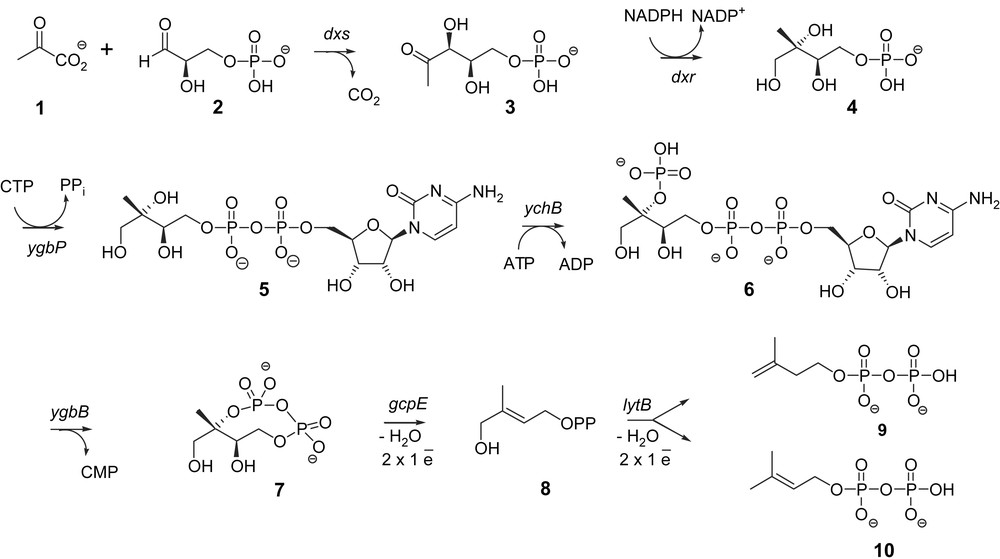
Methylerythritol 4-phosphate for isoprenoid biosynthesis.
2 Identification of gcpE and lytB as novel genes of the MEP pathway
To identify genes potentially involved in the MEP pathway, a bioinformatic approach was followed. Since bacterial genes with related functions are often organised in operons, uncharacterized open reading frames (ORFs) that are next to known genes of the MEP pathway were examined. An ORF with unknown function was found in the genome of Streptomyces coelicolor just upstream of the sequence coding for 1-deoxy-d-xylulose 5-phosphate synthase (DXS), the first enzyme of the MEP pathway. This ORF is homologous to the gcpE gene of Escherichia coli. Genes with homology to gcpE are broadly distributed in eubacteria and plants and the occurrence of the gcpE gene in completely sequenced bacterial genomes strictly correlates with the occurrence of the dxr gene-encoding 1-deoxy-d-xylulose 5-phosphate reductoisomerase (DXR), the second enzyme of the MEP pathway. To examine whether the gcpE gene is involved in the MEP pathway, this gene was deleted in an E. coli strain engineered for the synthesis of IPP (9) and DMAPP (10) from mevalonate. This means that in this strain the three genes of the mevalonate pathway required for the conversion of mevalonate into IPP were integrated in order to rescue the lethal mutations in the MEP pathway genes. The presence of mevalonate was required for the growth of this E. coli strain with a deleted gcpE gene, showing that gcpE is an essential gene in the MEP pathway [9].
The genomic occurrence of known genes of the MEP pathway correlates also with the occurrence of the lytB gene that was shown in a similar manner to be involved in isoprenoid biosynthesis [10]. Using again an E. coli strain capable of utilizing mevalonate lytB was shown to be an essential gene of the MEP pathway [11].
3 The (E)-4-hydroxy-3-methylbut-2-enyl diphosphate synthase (GcpE)
3.1 Identification of the reaction catalysed by the GcpE
The biological function of the GcpE protein encoded by the gcpE gene remained to be established. The strategy used for the tentative identification of the GcpE substrate was based on the construction of an E. coli strain capable of utilizing mevalonate, and harbouring simultaneously a disruption of the dxs gene, in order to improve the incorporation of free methylerythritol into isoprenoids [12,13], as well as a deletion of the gcpE gene, in order to favour a possible accumulation of the substrate of GcpE. This mutant was grown on LB medium containing mevalonate as well as [1-3H]methylerythritol and resulted in the accumulation of [1-3H]methylerythritol 2,4-cyclodiphosphate (7) that was the last intermediate described. This suggested that GcpE is the next enzyme of the MEP pathway [14].
GcpE from E. coli was cloned as a His-tagged protein and purified on an Ni2+ nitriloacetic acid agarose column. No activity was found when this enzyme was incubated with [2-14C]methylerythritol 2,4-cyclodiphosphate (7) and several cofactors. To overcome this lack of knowledge on the required cofactors and/or auxiliary enzymes, [2-14C]methylerythritol 2,4-cyclodiphosphate (7) was incubated with a crude cell-free system from an E. coli strain overexpressing GcpE. This crude cell-free system converted [2-14C]methylerythritol 2,4-cyclodiphosphate (7) into a new derivative, that was identified as (E)-4-hydroxy-3-methylbut-2-enyl diphosphate (8), only when the assay was performed using degassed buffers and working under an inert argon atmosphere [15,16]. This represented the first evidence that GcpE was an oxygen sensitive protein that converts methylerythritol 2,4-cyclodiphosphate (7) into HMBPP (8).
The GcpE catalysed reaction was simultaneously identified using an elegant in vivo approach [17]. Cyclodiphosphate (7) was accumulated from [U-13C6]-1-deoxy-d-xylulose by an E. coli strain that simultaneously overexpresses the gene of d-xylulose kinase, which phosphorylates DX to DXP (3), and the four genes of the MEP pathway, dxr, ygbP, ychB and ygbB (Fig. 1). Additional overexpression of the gcpE gene was followed by the formation of HMBPP (8), indicating that MEcDP (7) is the substrate and HMBPP (8) the reaction product of GcpE [17].
3.2 GcpE, an Fe–S protein
3.2.1 First evidences for the involvement of a metallic prosthetic group
The reaction catalysed by GcpE formally involves elimination and reduction steps. The reducing cofactor was, however, not identified, as no assay with the purified enzyme was available. In a bioinformatic search on the (I,V,L)(A,S)(V,I)(M,I,L)GCXVN(G,A)XGEXXX(A,S,T)XX (G,A) motif that is conserved in the GcpE orthologous group, a homology was found between this sequence and those of ferredoxins and aconitase, both of which are characterized by an active [4Fe–4S] cluster [16]. Pure GcpE from E. coli was therefore reconstituted using FeCl3 and Na2S in the presence of dithiothreitol in a glove box under a nitrogen atmosphere containing less than 2 ppm oxygen [18]. According to the assay conditions described for other enzymes with Fe–S cluster [19–21], reconstituted GcpE was tested either in the presence of the biological flavodoxin/flavodoxin reductase/NADPH reduction system or in the presence of 10-methyl-5-deaza-isoalloxazine (5-deazaflavin, DAF) semiquinone radical obtained by photoreduction. In the presence of either of these reduction systems, the reconstituted GcpE converted MEcDP (7) into HMBPP (8) whereas the non-reconstituted enzyme remained inactive [18]. One of the last bottlenecks in the elucidation of the MEP pathway has been solved: GcpE contains an Fe–S cluster.
3.2.2 Characterization of the [4Fe–4S] cluster of GcpE
The UV–vis spectrum of the khaki solution of the reconstituted enzyme displayed an absorption band at 410 nm, which is characteristic for proteins possessing a [4Fe–4S]2+ cluster [18]. Exposure of the reconstituted enzyme solution to air led to bleaching, corresponding to the fast degradation of the [4Fe–4S]2+ cluster to yield again the spectrum of the apoenzyme. Determination of the iron and the sulphur content revealed 4.4 iron ions and 4.9 sulphur atoms per reconstituted GcpE molecule, affording additional evidence for the presence of a [4Fe–4S] cluster.
The [4Fe–4S] prosthetic group of GcpE from E. coli and Arabidopsis thaliana was fully characterized by Mössbauer spectroscopy after reconstitution of the apoenzyme with 57Fe3+ chloride. It could be concluded that GcpE contains in a diamagnetic [4Fe–4S]2+ cluster three tetrahedrally sulphur-coordinated Fe2.5+ and one tetrahedrally coordinated Fe2.5+ with three sulphur ligands and one non-sulphur ligand [22]. Comparable Mössbauer spectroscopic signatures have also been found for the [4Fe–4S]2+ cluster of several enzymes such as the anaerobic ribonucleotide reductase from E. coli [23,24]. The spectroscopic assignment showed that the [4Fe–4S]2+ cluster is bound to the protein by three cysteine sulphurs and by an oxygen or a nitrogen of an unknown ligand that might be water, an amino acid or the substrate [22].
3.2.3 GcpE catalysis
The presence of a [4Fe–4S] cluster as a prosthetic group in GcpE suggested a radical reaction involving a reductive process with two one-electron transfers and radical intermediates (Fig. 2) [18,25]. The first cationic intermediate may also be replaced by an epoxide, as suggested by Arigoni and co-workers [25]. Alternatively, it was also proposed from modelling calculations that the second electron transfer occurs before the elimination of the C-3 hydroxy group [26]. The Lewis acid character of an Fe–S cluster, which may coordinate to a hydroxyl and facilitate its elimination, has been reported for a dehydratase by Buckel [27], but it is not known whether the Fe–S cluster and/or other acidic groups are involved.
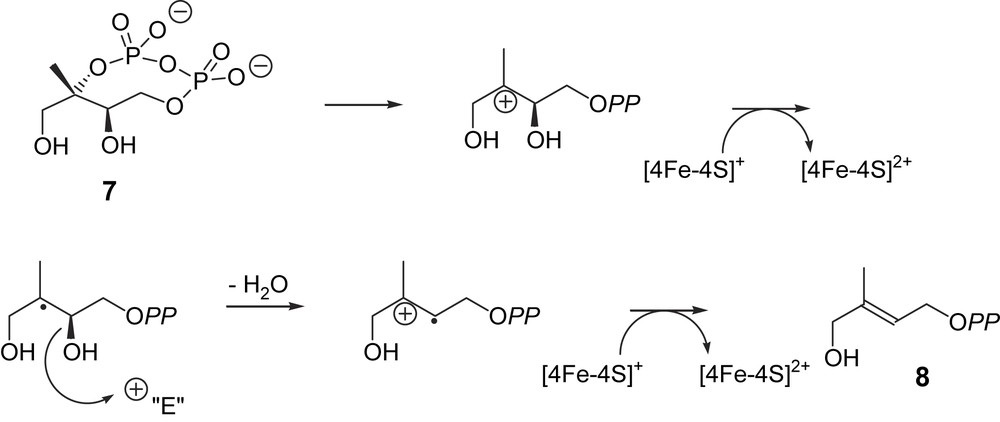
Hypothetical biogenetic pathway for the GcpE-catalysed reaction (intermediates are not free species but enzyme-bound stabilized entities) [18].
The catalytic activity of the reconstituted GcpE is close to 70 nmol min−1 mg−1 (M. Seemann, unpublished results). This activity is similar to that reported by the group of Bacher (45–100 nmol min−1 mg−1) for an as-isolated recombinant GcpE protein from E. coli purified under anaerobic conditions [28]. Actually, it was claimed in the latter work that the activity of this as-isolated enzyme was about two orders of magnitude higher than that of the reconstituted GcpE, for which an activity was intentionally not reported. A non-significant kcat was calculated for the reconstituted GcpE under non steady-state conditions, i.e. after a long incubation time chosen to accumulate as much reaction product as possible for its identification [18].
4 The (E)-4-hydroxy-3-methylbut-2-enyl diphosphate reductase (LytB)
The (E)-4-hydroxy-3-methylbut-2-enyl diphosphate reductase (LytB) was characterized by methods similar to those used for the identification of GcpE. In an E. coli mutant engineered for the utilization of exogenous mevalonate, HMBPP (8) accumulates upon deletion of the lytB gene and growth on mevalonate, affording the first evidence that HMBPP (8) is the substrate of the LytB enzyme [29].
In vivo experiments with recombinant E. coli strains showed that exogenous [U-13C6]-1-deoxy-d-xylulose can be converted into 13C-labeled IPP (9) and 13C-labeled DMAPP (10) by the consecutive action of the enzymes xylulose kinase, DXR, YgbP, YchB, YgbB, GcpE and LytB [30]. It was deduced that LytB was the last enzyme of the MEP pathway that catalysed the conversion of HMBPP (8) into IPP (9) and DMAPP (10) in a 5:1 ratio. As the LytB orthologous group contains three absolutely conserved cysteine residues, LytB was proposed to possess also an Fe–S prosthetic group [31]. Isolated LytB from E. coli was therefore reconstituted with FeCl3, Na2S and DTT under anaerobic conditions in a manner similar to the method previously described for GcpE [32]. LytB was tested in the presence of the biological flavodoxin/flavodoxin reductase/NADPH reduction system or in the presence of the DAF semiquinone radical obtained by photoreduction. Using either of these reduction systems, reconstituted LytB converted HMBPP (8) into IPP (9) and DMAPP (10) [32] whereas the as-isolated enzyme (which served for the reconstitution) was less active than the reconstituted LytB. LytB produced IPP (9) and DMAPP (10) in vivo or in isolated enzyme tests in an average ratio comprised between 4:1 and 6:1, depending on the origin of the enzymes (E. coli or Aquifex aeolicus) or on the conditions of the enzyme tests performed with as-isolated or reconstituted enzyme and with different associated reducing systems [30,32–34].
4.1 Characterization of the [4Fe–4S] cluster of LytB
Solutions of reconstituted LytB from E. coli are characterized by a khaki colour. The UV–vis spectrum displayed an absorption band at 410 nm, which is characteristic for proteins possessing a [4Fe–4S]2+ cluster [32]. The spectrum of the reconstituted enzyme did not significantly change after 30 min exposure to air at room temperature. Upon 4 h exposure, absorbance at 410 nm was still observed. In the same conditions, the absorbance of reconstituted GcpE nearly disappeared after 2 h. Determination of the iron and the sulphur content revealed 5 iron ions and 6 sulphur atoms per reconstituted lytB molecule, affording additional evidence for the presence of a [4Fe–4S] cluster [32]. These values, higher than the expected ones, are due to non-specific binding to the protein. After treatment of the reconstituted LytB with EDTA followed by desalting on a Sephadex G-25 column, reconstituted LytB contained 4.9Fe and 4S. The as-purified protein solution, which served for the reconstitution, was pale brown in colour and displayed an UV–vis spectrum with absorption characteristics reminiscent of an Fe–S cluster. Chemical analysis gave 0.8 iron and 0.6 sulphur atoms, respectively, per monomer.
Confirmation of the presence of a [4Fe–4S] cluster was obtained by EPR spectroscopy. Reconstituted LytB from E. coli is EPR-silent. This is in accordance with the presence of a [4Fe–4S]2+ centre. Reduction of the reconstituted enzyme with sodium dithionite induced the appearance of a signal with g values at g = 1.92(1) and 2.03(7), typical of an S = ½ [4Fe–4S]1+ cluster, to be compared to g values obtained by calculation (1.889, 1.927, 2.038) [32]. Microwave power saturation experiments on the reconstituted and dithionite reduced LytB at 20 K yielded a half-saturation power (P1/2) of 153 mW indicating the presence of a [4Fe–4S]1+ cluster. Quantification of the [4Fe–4S]1+ species by comparison with a Cu–EDTA standard signal, indicated a 72% reduction yield by sodium dithionite. In contrast, the as-purified enzyme gave an EPR signal with a g value at 2.02(1) (g values obtained by simulation: 1.956, 2.012 and 2.013), which is very similar to that of a [3Fe–4S]1+ centre. Microwave power saturation experiments yielded a P1/2 of 135 mW. Quantification of the signal showed that ca. 10% of the non-reconstituted LytB sample possessed a [3Fe–4S]1+ cluster. The combined results suggest that LytB initially contains a [4Fe–4S]2+ cluster, which degrades into the [3Fe–4S]1+ form in the presence of dioxygen during purification, a phenomenon frequently observed for [4Fe–4S]2+ proteins. Rohdich and co-workers presented an EPR spectrum of LytB from E. coli purified in anaerobic conditions and also concluded that LytB as-isolated in the absence of oxygen contains a [3Fe–4S]+ cluster [33]. In this latter experiment, the enzyme sample was not reduced, and the EPR signal not quantified. In such conditions, a [4Fe–4S]2+ cluster cannot be detected, even if it was present. In addition, the reported UV spectrum of this sample looked quite similar to the UV spectrum of reconstituted LytB. These observations on as-isolated LytB do not exclude that the active form of LytB possesses a [4Fe–4S]2+ cluster and that the [3Fe–4S]+ form results from the degradation of the former.
4.2 The LytB catalysed reaction
LytB reduces HMBPP (8) into a mixture of IPP (9) and DMAPP (10). The requirement for a [4Fe–4S] cluster for the LytB activity strongly suggests that this prosthetic group is involved in a redox process, i.e. the reduction occurs via two successive one-electron transfers from the reduced [4Fe–4S]1+ form, finally yielding, probably via radical intermediates, the allylic anionic intermediate (11), which is protonated either at C-2 yielding IPP (9) or at C-4 yielding DMAPP (10) (Fig. 3) [25,35]. The enantioselectivity of the protonation of intermediate (11) can be deduced from former results on the MEP pathway. According to the labelling pattern observed in the prenyl chains of ubiquinone and menaquinone of E. coli, after feeding of [4-2H]deoxyxylulose or [3-2H]methylerythritol, the hydrogen atom at C-4 of DXP (3) or at C-3 of MEP (4) is retained in the isoprene units derived from DMAPP (10) and lost in those derived from IPP (9) [36,13]. This observation is consistent with the retention of this hydrogen atom through the whole MEP pathway, i.e. up to HMBPP (8) and the anionic intermediate (11), most probably yielding [2-2H]DMAPP and [2R-2H]IPP (Fig. 3). Indeed, in the chain elongation, the prenyl transferase eliminates the pro-R hydrogen of IPP and retains the pro-S hydrogen. Accordingly, the protonation of the anionic allylic intermediate (11) has to occur from the si face (Fig. 3).
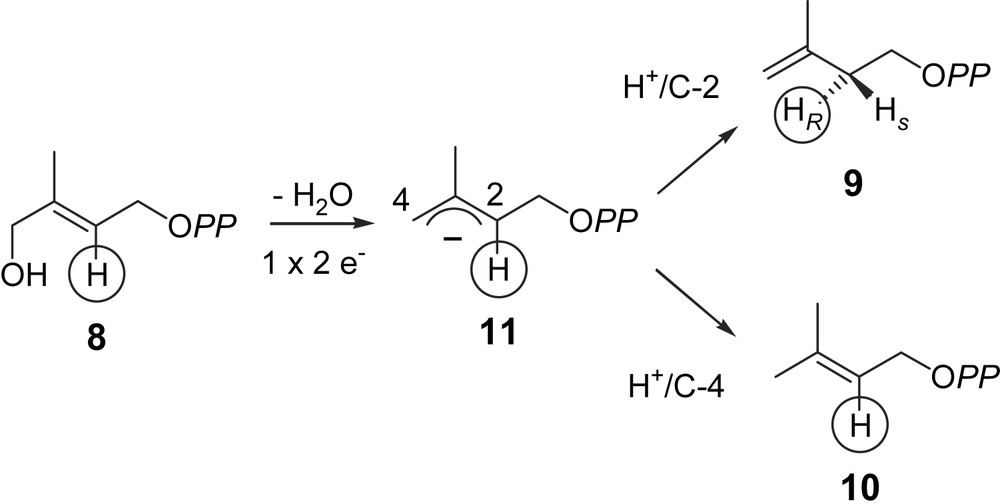
Hypothetical mechanism for the LytB catalysed reaction [25,32,34].
5 The reduction system of the [4Fe–4S]2+ cluster of GcpE and LytB
For catalytic activity, GcpE and LytB have to be coupled with a system allowing the reduction of the oxidized [4Fe–4S]2+. This can be completed in the bacterium E. coli by the natural flavodoxin/flavodoxin reductase/NADPH system [18,25]. The fldA gene encoding flavodoxin I was found to be essential in E. coli and supposed to be mainly involved in the reduction of the GcpE and LytB Fe–S clusters [37]. Reduction can also be performed by chemical means: e.g. with the semiquinone radical of 5-deazaflavin (DAF) for the recombinant GcpE enzyme from E. coli [18] and A. thaliana [22] or even dithionite for GcpE from the bacterium Thermus thermophilus [38] and LytB from A. aeolicus [34]. GcpE from the plant A. thaliana showed, however, no activity in the presence of the flavodoxin/flavodoxin reductase from E. coli and NADPH [22]. In addition, flavodoxin is absent in the plastids of phototrophic organisms and in the plastid-derived apicoplasts of the Plasmodium spp. [39,40]. The electron transfer from ferredoxin onto GcpE is apparently possible in the cyanobacterium Thermosynechococcus elongatus lacking flavodoxin [41]. LytB from E. coli displays some reducing activity in the presence of spinach ferredoxin/ferredoxin-NADP+ reductase/NADPH [30], whereas the LytB from P. falciparum was shown to be catalytically active in the presence of the same reduction system [40]. In contrast, the plant GcpE from A. thaliana is fully active in the presence of spinach thylakoids, upon illumination and in the absence of NADPH [42]. This shows that the electron flow from photosynthesis can directly provide via ferredoxin the electrons required for the GcpE catalysed reaction, without any contribution of the ferredoxin-NADP+ reductase and NADPH, the latter energy-consuming system being only required in roots or in photosynthesising tissues in the dark (Fig. 4) [42].
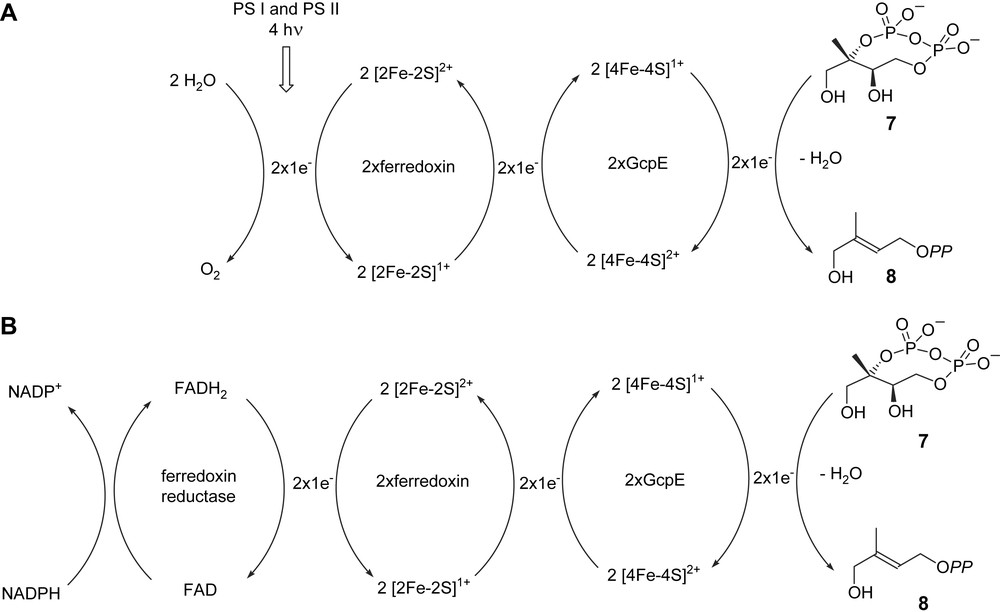
Hypothetical biogenetic scheme for plant GcpE activity: (A) in photosynthetic tissues in the light; (B) in photosynthetic tissues in the dark and in non-photosynthetic tissues. PS I and PS II: photosystems I and II [42].
6 Concluding remarks
One of the reasons the MEP pathway was overlooked for many decades is the oxygen sensibility of the last two enzymes, possessing both a [4Fe–4S]2+ cluster. Even if the mode of action of GcpE and LytB is now roughly delimitated, many unsolved are pointed out. Their [4Fe–4S] cluster was characterized in isolated enzymes either directly after isolation, or upon reconstitution. What is the prosthetic group in vivo? This question could be answered by Mössbauer spectroscopy on bacterial or plant cells grown in the presence of a 57Fe salt. The associated reducing systems are different in E. coli (flavodoxin) and in plants (ferredoxin), at least in tests performed with the purified enzyme. What is (are) the reducing system(s), which is (are) involved in vivo, in normal growth conditions? In plants, the O2 sensitive GcpE and LytB are located in the chloroplasts, i.e. in a site producing O2 in phototrophic conditions. How are the active sites of these enzymes protected against O2 damage? Are protecting and/or reparation proteins involved? This raises also the question of the biosynthesis of the cluster in the active site. The intervention of proteins of the isc operon has been suggested [33]. Finally, radical intermediates have been postulated for the reactions catalysed by GcpE and LytB. Attempts should be made for identifying them by EPR spectroscopy. This would give a complete overview on these reactions.
The MEP pathway is absent in animals, yeast and fungi, but present in most bacteria, including pathogens as well as in P. falciparum responsible for malaria. The MEP pathway is therefore an interesting target for the design of novel antibacterial and antiparasitic drugs, with minimal side effects for the patient [43]. Even if most bacterial species do not produce large amounts of isoprenoids, these metabolites are essential. Any inhibition of their biosynthesis is lethal.
Acknowledgments
The authors are indebted to Prof. A. Marquet, Dr. B. Tse Sum Bui, Prof. A. Boronat, Dr. M. Miginiac-Maslow and Prof. V. Schünemann for numerous valuable discussions. This work was supported by grants from the ‘Institut universitaire de France’ to M.R., and from the ‘Agence nationale de la recherche’ (grant No. ANR-05-JCJC-0177-01) and from the PROCOPE Germany/France Joint Research program to M.S.