

1 Introduction
The field of artificial metalloenzymes, based on the incorporation of an active catalyst, in a macromolecular host has enjoyed a revival in recent years [1,2]. Both covalent and supramolecular anchoring strategies have successfully been exploited to produce enantioselective hybrid catalysts for ester hydrolysis [3], dihydroxylation [4], epoxidation [5], sulfoxidation [6–11], hydrogenation [12–19], transfer hydrogenation [20,21] and Diels–Alder reactions [22–24].
Based on Whitesides' early report [25], we have been exploiting the biotin–avidin technology to produce artificial hydrogenases for the enantioselective reduction of N-protected dehydroamino acids [13,14,18,19] as well as the reduction via transfer hydrogenation of aromatic ketones [20,21]. For optimization purposes, we rely both on chemical- and genetic-strategies (i.e. chemogenetic) to yield both (R)- and (S)-selective hydrogenases. For the proof-of-principle, we focused exclusively on achiral ligands and spacers, thus ensuring that (enantio)selectivity is provided by second coordination sphere between the catalyst–substrate and the host protein. Following this chemogenetic optimization, good enantioselectivities (>90% ee) for both enantiomers of N-protected amino acids (N-acetamidoalanine N-AcAla and N-acetamidophenylalanine N-AcPhe) could be obtained using streptavidin (WT-Sav hereafter) as host protein. Herein we report on our efforts to produce active and selective artificial hydrogenases based on the biotin–avidin technology, with an emphasis on the chemical optimization using enantiopure α-amino acids (Scheme 1). A preliminary account of this work has appeared recently [19].
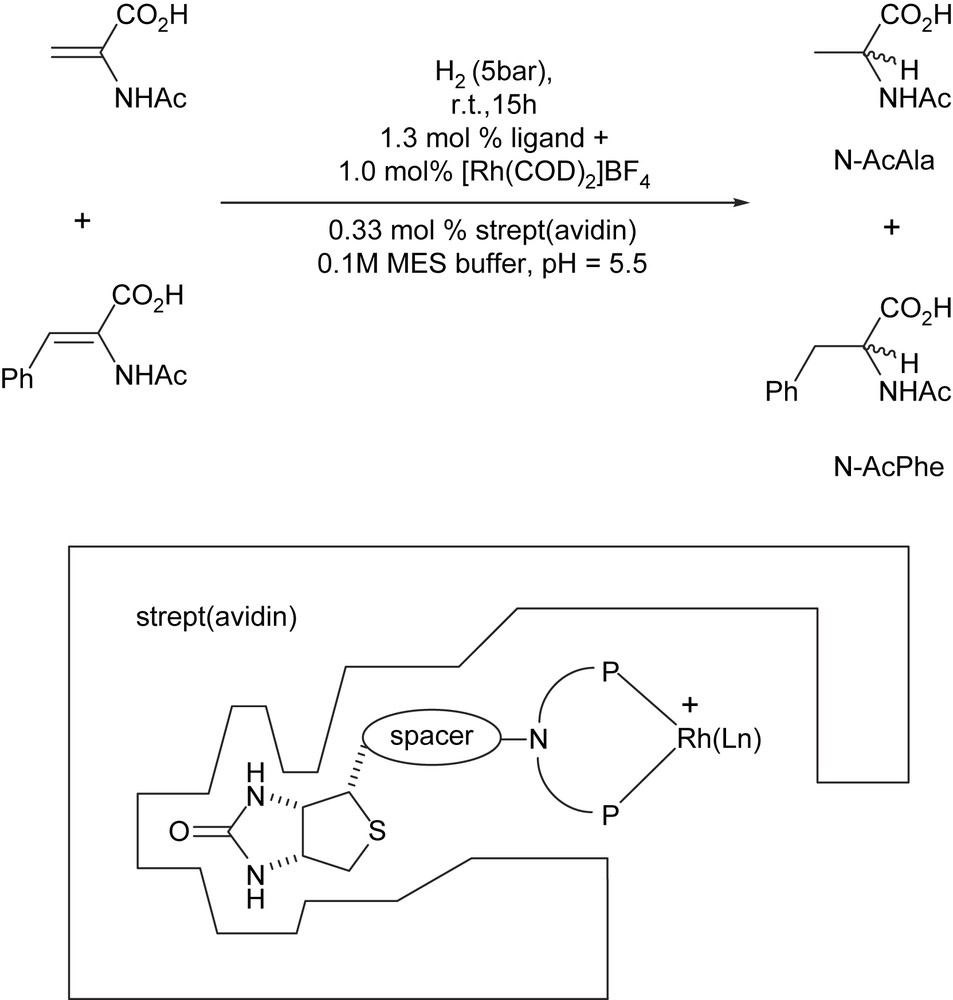
Artificial hydrogenases based on the biotin (strept)avidin technology for the reduction of N-protected dehydroamino acids.
2 Results and discussion
2.1 Choice of ligand and spacer
In previous studies [13,14,18], we demonstrated that the nature of the aminodiphosphine moiety (H-NP2) plays a critical role in both the activity and the selectivity of the resulting artificial metalloenzymes. Although the diphosphine 1 proved superior in most cases, the diphosphine 2 displayed interesting substrate- and enantioselectivities in a few cases [14]. We thus included both scaffolds 1 and 2 and combined them with enantiopure amino acid spacers (aa) to afford Biot-aa-NP2. For these studies, we selected phenylalanine ((R)-Phe, (S)-Phe) and proline ((R)-Pro, (S)-Pro) as spacer to afford a total of eight biotinylated ligands, Scheme 2. The choice of spacer was influenced by the fact that the biotin-binding pockets in both Sav and Avi are hydrophobic; we thus speculated that the presence of an aromatic moiety on the spacer could favorably interact with the tryptophane (and phenylalanine in WT-Avi) residues present in the binding pocket. Proline clearly stands out among the natural amino acids; we reasoned that introduction of pyrrolidine ring as a spacer would restrict the degrees of freedom of the catalyst within streptavidin (catalyst ⊂ WT-Sav; the inclusion symbol “⊂” will be used throughout this section to describe artificial metalloenzymes: metal fragment ⊂ host protein).
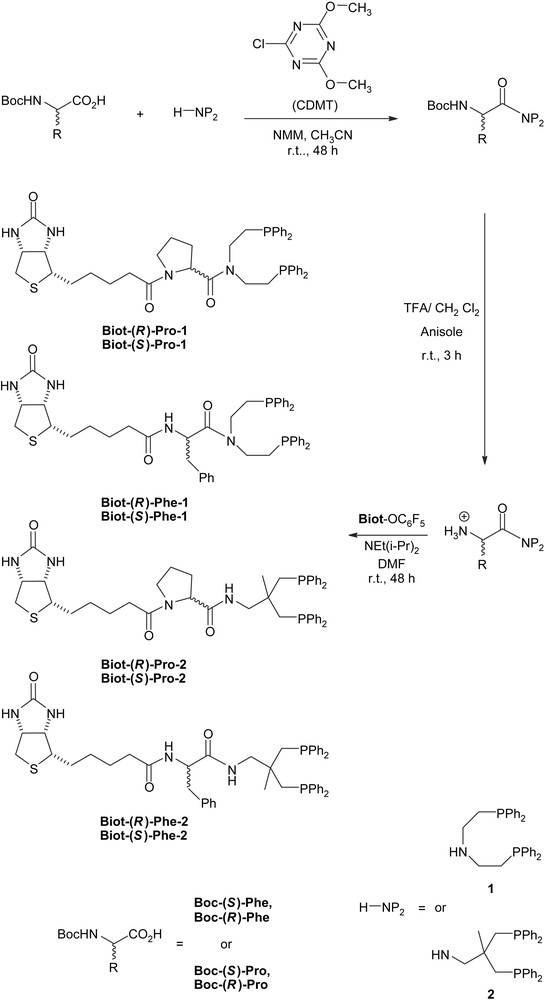
Synthesis of biotinylated diphosphine ligands incorporating chiral spacers.
2.2 Ligand synthesis
In the spirit of peptidic coupling with enantiopure amino acids and in order to avoid epimerization upon amide bond formation, we opted for an N → C coupling scheme, rather than the C → N approach used previously with achiral amino acid spacers, (Scheme 2) [13,18].
The aminodiphosphine scaffold H-NP2 1 or 2 was coupled with the Boc-protected amino acids activated in situ with CDMT (chlorodimethoxytriazine) [26]. The yields of the purified product critically depend on the diphosphine moiety: 60–70% yields were obtained with the diphosphine 1, whereas with the sterically congested, neopentylic amine 2, they only reach 20–30% [19].
The deprotection of the amino group was achieved with trifluoroacetic acid in the presence of two equivalents of anisole to avoid attack of the tert-butyl cation on the aromatic groups at phosphorus [27].
The crude ammonium salts were reacted with activated biotin, namely (+)-biotin-pentafluorophenylester [28], under basic conditions. After chromatography on silica gel under inert atmosphere, the desired ligands were isolated as white to pale yellow oils in 5–41% overall yield (three steps). Next, we proceeded to test these eight ligands with WT-Sav in their performance as artificial hydrogenases. All experimental details concerning the synthesis and characterization of all intermediates and ligands can be found in the supporting information of our preliminary communication [19].
2.3 Screening results
As the biotinylated ligands bear an enantiopure amino acid moiety relatively close to the chelating diphosphine moiety, we checked their performance, both in terms of activity and of selectivity in the hydrogenation of α-acetamidoacrylic acid in the absence of protein (Table 1).
Results for the hydrogenation of α-acetamidoacrylic acid in the absence of protein
| Entry | Ligand | ee (%) N-AcAla | Conversion (%) |
| 1 | Biot-1 | 0 | 81 |
| 2 | Biot-(S)-Phe-1 | 7 (S) | 100 |
| 3 | Biot-(R)-Phe-1 | 3 (R) | 91 |
| 4 | Biot-(S)-Phe-2 | 7 (S) | 92 |
| 5 | Biot-(R)-Phe-2 | 2 (R) | 100 |
| 6 | Biot-(S)-Pro-1 | 8 (R) | 100 |
| 7 | Biot-(R)-Pro-1 | 3 (R) | 100 |
| 8 | Biot-(S)-Pro-2 | 8 (S) | 100 |
| 9 | Biot-(R)-Pro-2 | 0 | 100 |
Catalysis reactions were run in degassed (nitrogen bubbling) MES buffer (0.1 M, pH = 5.5, MES = 2-(N-morpholino)ethanesulfonic acid). To ensure solubility, the rhodium complexes were prepared in situ in DMSO, which, upon mixing with the protein and the substrate, accounts for 9% of the total volume. Experimental details, including catalytic setup and GC operating conditions can be found in Ref. [14].
All ligands afford high conversion rates and, not unexpectedly, low enantioselectivity: a maximum of 8% ee in favor of the (R)-N-AcAla, entry 7, and (S))-N-AcAla, entry 9. We next tested these ligands in the hydrogenation of a cocktail containing both α-acetamidoacrylic and α-acetamidocinnamic acid in the presence of WT-Sav.
The results displayed in Table 2 suggest that the ligands carrying the diphosphine moiety 1, Biot-aa-1, perform better than the corresponding Biot-aa-2, both in terms of activity and of selectivity. This general feature emphasizes once again the critical role of the ligand structure on the performance of the artificial metalloenzyme. Hydrogenations of α-acetamidocinnamic acid with hybrid catalysts derived from Biot-aa-2 yield modest enantioselectivities (up to 60% ee) and modest conversions. In contrast, with ligands based on diphosphine 1, Biot-aa-1, good conversions and good selectivities (up to 91% ee) are found. The most efficient ligand is derived from unnatural (R)-Proline, Biot-(R)-Pro-1, which affords full conversion in each case and up to 91% ee (S) for N-AcPhe and 86% ee (S) for N-AcPhe in the presence of WT-Sav.
Results for the hydrogenation of α-acetamidoacrylic acid and α-acetamidocinnamic acid (as a mixture) in the presence of [Rh(Biot-aa-NP2)(COD)]+ ⊂ WT-Sav
| Entry | Ligand | N-AcAla | N-AcPhe | ||
| ee (%) | Conv. (%) | ee (%) | Conv. (%) | ||
| 1 | Biot-(R)-Pro-1 | −86 | 100 | −91 | 100 |
| 2 | Biot-(S)-Pro-1 | 23 | 100 | 23 | 100 |
| 3 | Biot-(R)-Phe-1 | 66 | 100 | 64 | 100 |
| 4 | Biot-(S)-Phe-1 | −73 | 100 | −64 | 87 |
| 5 | Biot-1 | 94 | 100 | 93 | 84 |
| 6 | Biot-(R)-Phe-2 | 5 | 100 | 6 | 68 |
| 7 | Biot-(S)-Phe-2 | −56 | 100 | −16 | 37 |
| 8 | Biot-(R)-Pro-2 | 9 | 100 | −14 | 54 |
| 9 | Biot-(S)-Pro-2 | −40 | 100 | −32 | 73 |
| 10 | Biot-2 | −6 | 38 | 0 | 2 |
Depending on the absolute configuration of the Phe spacer, nearly identical, but opposite enantioselectivities (>60% ee) are obtained for [Rh(Biot-Phe-1)(COD)]+ ⊂ WT Sav, Table 2, entries 3 and 4.
It appears that the amino acid spacer interacts very significantly with the protein environment, thus dramatically influencing the position of the Rh moiety within the chiral environment provided by the host protein.
Having created (S)-selective artificial hydrogenases with WT-Sav as host protein, we proceeded to evaluate the solvent tolerance of [Rh(Biot-(R)-Pro-1)(COD)]+ ⊂ WT-Sav. For this purpose, catalytic runs were performed in the presence of increasing amounts of different organic solvents. For comparison, we use [Rh(Biot-1)(COD)]+ ⊂ WT-Sav, which was previously shown to be a versatile (R)-selective hybrid catalyst [13,14,18]. In order to ensure solubilisation of the catalyst precursor, typical catalytic runs are performed with a buffer:DMSO ratio of 91:9, which was used as a starting point.
The results of the catalytic runs with [Rh(Biot-1)(COD)]+ ⊂ WT-Sav and [Rh(Biot-(R)-Pro-1)(COD)]+ ⊂ WT-Sav in the presence of 9 to 45% dimethylsulfoxide are collected in Table 3.
Catalytic runs with Biot-1 and Biot-(R)-Pro-1 in the presence of increasing amounts of DMSO
| Entry | % DMSO | Ligand | N-AcAla | N-AcPhe | ||
| ee (%) | Conv. (%) | ee (%) | Conv. (%) | |||
| 1 | 9 | Biot-1 | 94 | 100 | 94 | 85 |
| 2 | 9 | Biot-(R)-Pro-1 | −87 | 100 | −91 | 100 |
| 3 | 18 | Biot-1 | 90 | 100 | 88 | 90 |
| 4 | 18 | Biot-(R)-Pro-1 | −87 | 100 | −91 | 100 |
| 5 | 27 | Biot-1 | 81 | 100 | 80 | 80 |
| 6 | 27 | Biot-(R)-Pro-1 | −87 | 100 | −91 | 100 |
| 7 | 36 | Biot-1 | 52 | 80 | 60 | 58 |
| 8 | 36 | Biot-(R)-Pro-1 | −87 | 100 | −87 | 100 |
| 9 | 45 | Biot-1 | 15 | 72 | 23 | 25 |
| 10 | 45 | Biot-(R)-Pro-1 | −87 | 100 | −87 | 96 |
With Biot-1 ⊂ WT-Sav, we observe a significant erosion of enantioselectivity (and to a lesser extent conversion), which drops to 15% ee N-AcAla and to 23% ee for N-AcPhe in the presence of 45% DMSO. From these data, it is clear that the Biot-1-based catalytic system does not tolerate large amounts of DMSO.
In strong contrast, hybrid catalysts derived from Biot-(R)-Pro-1 tolerate large amounts of DMSO. Indeed, only a very modest erosion of enantioselectivity is observed.
Other organic co-solvents (9%) were tested in the presence of Biot-(R)-Pro-1 ⊂ WT-Sav. Ethanol, methanol, acetone, tetrahydofuran and dimethylformamide were selected, and the results are summarized in Table 4.
Catalytic runs with Biot-(R)-Pro-1 in the presence of 9% (in volume) of various organic co-solvents
| Solvent (9% in volume) | N-AcAla | N-AcPhe | ||
| ee (%) | Conv. (%) | ee (%) | Conv. (%) | |
| DMSO | −87 | 100 | −91 | 100 |
| EtOH | −86 | 100 | −86 | 100 |
| MeOH | −81 | 100 | −83 | 100 |
| Acetone | −81 | 100 | −82 | 100 |
| DMF | −87 | 100 | −88 | 100 |
| THF | −38 | 38 | −44 | 35 |
Except for THF, which is recognized as a strongly denaturing solvent, both the activity and the enantioselectivity is by and large maintained with the other water-miscible solvents tested. Interestingly, dimethylformamide, which is seldom used for enantioselective hydrogenation of enamides with cationic rhodium due to its coordination ability, is compatible with artificial hydrogenases. These results consolidate our findings that the catalytic system based on the Biot-(R)-Pro-1 ligand is indeed robust. This prompted us to evaluate further its performance under biphasic reaction conditions (Table 5). For substrate solubility reasons, ethylacetate was selected as non-miscible solvent and used as a 1:1 biphasic mixture combined with MES buffer.
Biphasic catalytic runs with [Rh(Biot-(R)-Pro-1)(COD)]+ ⊂ WT-Sav in the presence of 50% ethylacetate
| Catalyst | N-AcAla | N-AcPhe | ||
| ee (%) | Conv. (%) | ee (%) | Conv. (%) | |
| [Rh(Biot-(R)-Pro-1)(COD)]+ ⊂ WT-Sav | −83 | 96 | −88 | 90 |
| [Rh(Biot-1)(COD)]+ ⊂ WT-Sav | 30 | 56 | 31 | 5 |
Under biphasic conditions, [Rh(Biot-(R)-Pro-1)(COD)]+ ⊂ WT-Sav, performs much better than its [Rh(Biot-1)(COD)]+ analog.
3 Conclusion
The straightforward incorporation of enantiopure amino acid spacers into biotinylated diphosphine ligands allowed us to identify an efficient artificial hydrogenase based on the biotin–avidin technology. The catalytic system [Rh(Biot-(R)-Pro-1)(COD)]+ ⊂ WT-Sav displays unprecedented (S) selectivity (up to 91% ee in streptavidin) and enhanced stability towards organic co-solvents. It therefore broadens the scope of artificial metalloenzymes based on the biotin–avidin technology. Its efficiency in biphasic medium opens the way to new applications in the asymmetric hydrogenation of water insoluble prochiral alkenes with artificial metalloenzymes and recycling of the water soluble catalyst.
Acknowledgments
We thank Prof. C. R. Cantor for the streptavidin gene as well as Profs. P. Schürmann and J.-M. Neuhaus for their help in setting up the protein production. This work was funded by the Swiss National Science Foundation (Grants FN 620-57866.99, and FN 200021-105192/1 as well as NRP 47 “Supramolecular Functional Materials”), CERC3 (Grant FN20C321-101071), the Roche Foundation, the Canton of Neuchâtel as well as FP6 Marie Curie Research Training Network (IBAAC network, MRTN-CT-2003-505020). Umicore Precious Metals Chemistry is acknowledged for a generous loan of rhodium.