

1 Introduction
In the recent years, a huge research effort has been devoted to the design of artificial metalloenzymes that are able to perform efficiently, under mild conditions, regio- and stereoselective oxidation reactions [1]. This growing interest has mainly been driven by the fact that such biocatalysts will find numerous applications in oxidation processes with considerable economic potential, and wherever clean oxidations under mild conditions are needed: gas and petroleum industry, decontamination and detoxification of industrial wastes, or fine chemistry and oxidation of molecules of pharmaceutical interest.
Nature has already solved the problem of the selective oxidation of substrates under particularly mild conditions, as it uses as oxidant the most abundant, cheapest and cleanest one as possible, dioxygen, in the presence of metalloenzymes as catalysts at room temperature under atmospheric pressure. Indeed, in the biological world, metal-containing proteins are able to perform oxidation reactions under particularly mild conditions, even the hydroxylation of hydrocarbons, in spite of the relative inertness of the C–H bond in non-activated substrates. These include non-heme enzymes, such as methane monooxygenase, which is able to catalyze the oxidation of methane into methanol, and heme enzymes, such as cytochrome P450-dependent monooxygenases.
As a consequence, in the last 10 years, several strategies have then been reported for the design of artificial metalloproteins that are able to perform such oxidation reactions and use if possible dioxygen as oxidant. These strategies have been developed either to increase or to modify the reactivity of an already existing metalloprotein, or to induce a new reactivity in a non-metal protein. They include the introduction of metal binding sites in the active site of a protein, by site-directed mutagenesis [2–7], design of substrate binding cavities [8–11], chemical modification of prosthetic groups [12–15], and covalent attachment of metal cofactors [16–20].
Since we had long been involved in the study of the reactivity of hemoproteins and the design of biomimetic systems involving synthetic metalloporphyrins, we chose to build up new artificial hemoproteins which would associate monoclonal antibodies with synthetic metallotetraarylporphyrins. The present paper reviews the different strategies we have used and the results obtained compared with those of other groups in the world.
2 Requirements for the elaboration of biomimetic systems for monooxygenase-like hemoproteins
Two main questions arise for the elaboration of biomimetic systems for metalloenzymes and more specifically for monooxygenase-like hemoproteins. The first one is with regard to the structure–activity relationships, which structural elements are indispensable in the biomimetic system? The second one is which oxidant should we choose, dioxygen, like in natural systems, or other oxidants such as iodosylbenzene, organic or inorganic peroxides? Before we answer those questions, we need to have a look at structure–activity relationships in cytochrome P450s-dependent monooxygenases.
For historical reasons, the superfamily of the cytochrome P450s, enzymes that hydroxylate substrates according to reaction (1), still remains the most investigated class of metalloenzymes involved under atmospheric oxidation [21]. Cytochrome P450s are ubiquitous enzymes but a large proportion of them are mainly located in hepatocytes where they are able to oxidize a wide range of endogenous as well as exogenous substrates, such as drugs and pollutants.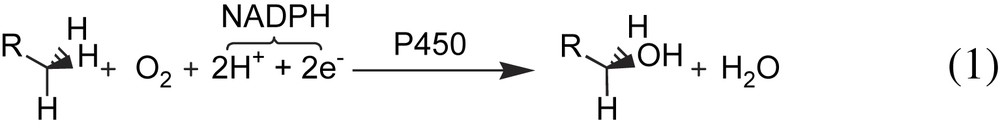
Their active site is a wide hydrophobic pocket which contains the prosthetic group, a heme b, which is bound to the apoprotein through a cysteinate axial ligand of the iron [22,23], and a binding site for the substrate (Fig. 1).
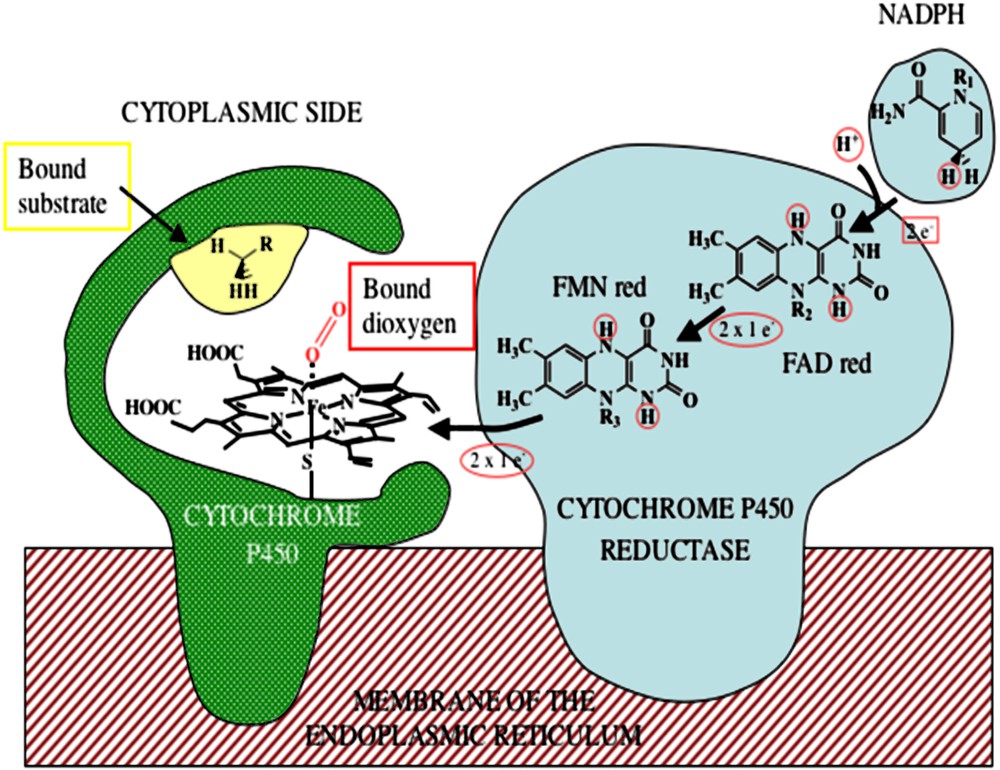
Electron transfer through the cytochrome P450–cytochrome P450 reductase complex.
The apoprotein plays then at least three key roles: it first controls the positioning of the substrate with respect to the heme, inducing the stereoselectivity of the reaction, it modulates the redox potential of the heme, and it protects the heme from oxidative degradations.
In the reactions catalyzed by cytochrome P450s, dioxygen is activated in the presence of two protons and two electrons that are provided by a cellular reductant NADPH, and one oxygen atom is reduced to water, the other being transferred into the substrate (reaction (1)). The catalytic cycle of the P450s is now well established [22,23]. It involves reduction of the metal, from ferric to the ferrous state, binding of molecular dioxygen to the metal, further reduction which leads in protic medium to the formation of the so-called “active oxygen species”, a high-valent iron-oxo species responsible for the hydroxylation of the substrates (Scheme 1). The nature of this powerful oxidizing intermediate can be deduced from cross-linked investigations using a broad array of spectroscopic techniques [24,25].
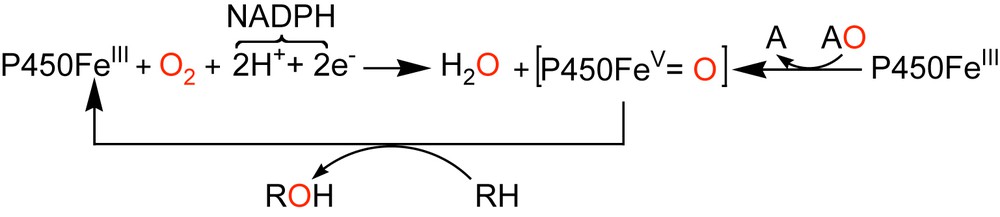
Formation of the high-valent iron-oxo species involved in the catalytic cycle of Cyt P450.
Relatively simple metal-containing active sites can be found in many enzymes from many sorts of microorganisms. In these sophisticated molecular machines, the enzymes whose function is to perform important oxidation reactions work in the presence of molecular dioxygen and a source of energy: electrons delivered through specific sources like reductase proteins for instance, or biological two-electron donating cofactors such as tetrahydrobiopterins [26,27]. In the particular case of cytochrome P450s, the electrons necessary for the reduction of dioxygen are not directly transferred from NADPH to the iron center, but they are delivered one by one by the cytochrome P450-reductase, a huge protein which contains two flavin cofactors, FAD and FMN.
It is noteworthy that, in vitro, high-valent iron-oxo species can directly be obtained, by the so-called “peroxide shunt”, from the reaction of iron(III)-P450 with various oxygen donating agents (AO) such as iodosylbenzene (PhIO), hydrogen peroxide (H2O2), alkylhydroperoxides (ROOH) or inorganic oxidants such as sodium hypochlorite (NaOCl), oxone (KHSO5) [28].
Back to the questions that we raised previously, considering the aforementioned structure–activity relationships in hemoproteins, at least three structural elements have to be mimicked to elaborate the ideal functional biomimetic system for cytochrome P450-dependent monooxygenases (Fig. 1): (i) the prosthetic group, a heme or iron(III)-protoporphyrin IX, which is responsible for the oxene-transfer reactions [29,30]; (ii) the apoprotein which binds and site-isolates heme, preventing its aggregation and its oxidative degradation, and its active-site amino acids, which provide an hydrophobic environment for the substrate and control its access to the heme, and (iii) the proximal axial ligand of the iron atom, a cysteinate, which has a role in controlling the heme redox potential and the reactivity of the high-valent iron-oxo intermediates involved in their catalytic cycle [31].
Concerning the choice of the oxidant, as we mentioned above, the use of dioxygen requires a sophisticated machine to transfer the electrons to the iron atom to activate dioxygen. On the contrary, since (i) the “in vitro” short cycle only requires the one-pot association of cytochrome P450 and single oxygen atom donors (AO), and (ii) model systems, simply associating an O atom donor and an iron(III)- or manganese(III)-porphyrin, have been found to be able to perform all the reactions typical of cytochrome P450, without the need for an axial ligand of the metal to mimic the cysteinate proximal ligand of heme in cytochrome P450 [29,32], it thus seemed much easier to design biomimetic systems that used single oxygen atom donors (AO) as oxidant.
3 Design of new biocatalysts based on monoclonal antibodies: towards a new generation of artificial hemoproteins
The production of monoclonal antibodies raised against transition state analogs has proven to be a powerful strategy to obtain antibodies that are able to catalyze a wide range of reactions [33–44]. However, since most of these catalytic antibodies have modest catalytic efficiencies, several other strategies have been envisioned. One of them is based on the association of antibodies with cofactors such as inorganic cofactors [45,46], natural cofactors [47], metal ions [48–50], or metal cofactors [52–78]. In this strategy, the antibodies are designed either to bring into close proximity the cofactor and the substrate, or to bind tightly the cofactor to enhance its reactivity. Thus, antibodies with metalloporphyrin cofactor appeared as an alternative approach to provide a route to catalysts tailored for specific oxidation reactions that would fulfill the above-mentioned requirements to elaborate the ideal functional biomimetic system for Cyt P450-dependent monooxygenases. The main difficulty thus consisted in designing the ideal porphyrin hapten, which could induce in the paratope of the antibody, not only a binding site for the metalloporphyrin, but also a hydrophobic pocket to accommodate the substrate and, if possible, an axial ligand of the iron.
Numerous teams have faced this problem in the last 15 years, and they have raised antibodies against porphyrins, either natural heme derivatives (Fig. 2A) or synthetic tetraarylporphyrins (Fig. 2B) in order to get porphyrin–antibody complexes having cytochrome P450-like or peroxidase-like activities.
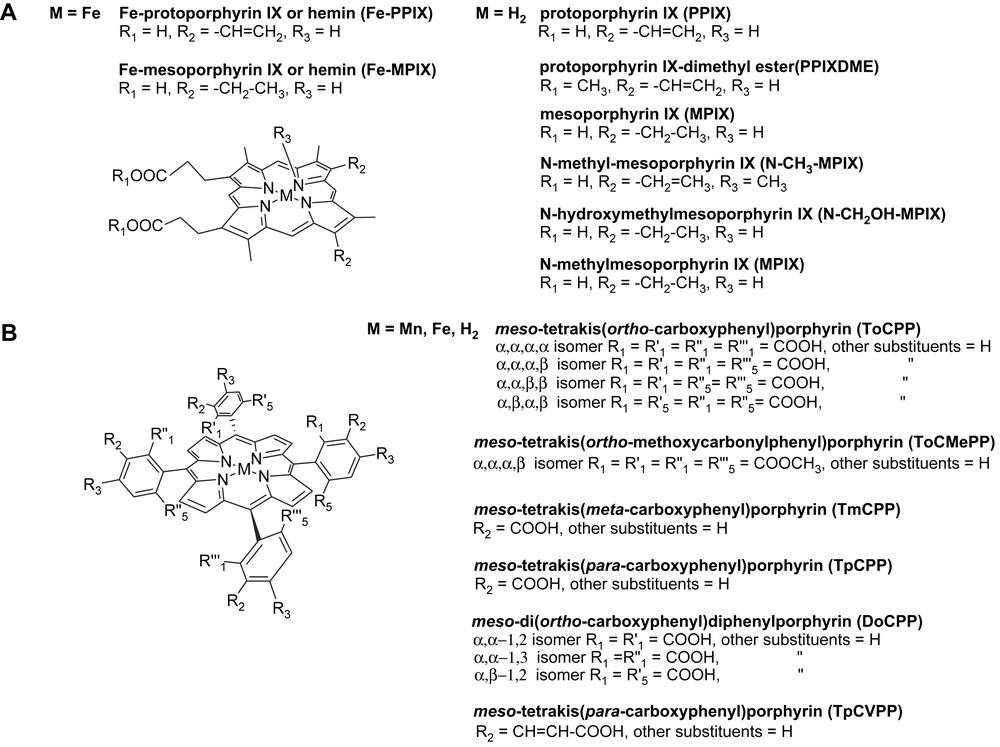
Structure and nomenclature of the various porphyrins mentioned in this article.
We first thought about raising antibodies against synthetic capped hemes, bearing an analog of substrate above the plane of the porphyrin, in order to raise bi-site antibodies that would possess a binding site for a metalloporphyrin cofactor and above a second binding site to accommodate selectively the substrate (Fig. 3).
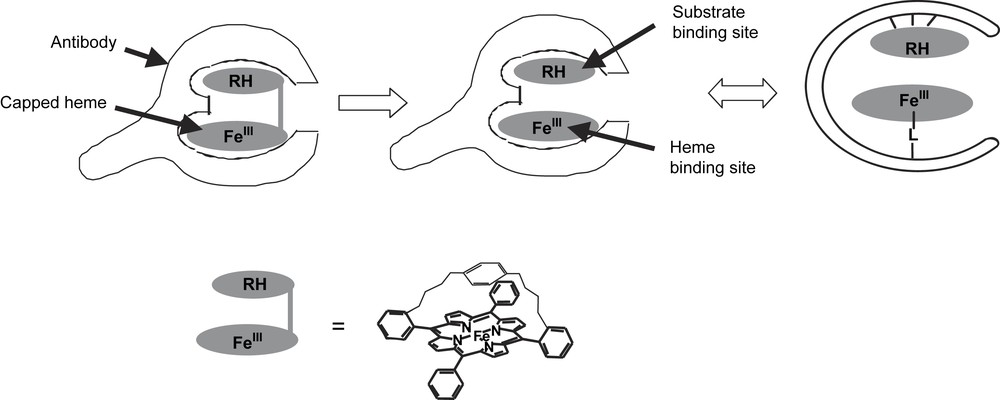
Elaboration of antibody–porphyrin complexes with a monooxygenase-like activity: production of bi-site antibodies.
Unfortunately, these bulky aromatic molecules appeared to be poorly immunogenic and the few antibodies generated were IgMs that displayed a weak affinity for the iron-porphyrin (Kd = 10−4 M). In addition, no catalytic activity was found for the corresponding Fe-porphyrin–antibody complexes. We thus turned to another strategy inspired from the structure–activity relationships in heme peroxidases.
3.1 Antibody–porphyrin complexes with a peroxidase-like activity
The active site of peroxidases, which contains a heme bound to the apoprotein by a histidine residue, is relatively narrow and little space is left over the plane of the porphyrin, so that only the oxidant, H2O2 or ROOH, can enter the active site and interact with the iron atom, whereas the reducing co-substrates only interact with the edge of the heme [79,80]. Thus, at first sight, peroxidases appeared to be some of the easiest hemoproteins to be mimicked, and to induce the production of antibodies having a binding site with such characteristics, one simple strategy was to use as haptens quite unsophisticated flat porphyrins.
In the mechanism of heme peroxidases, the key step is the heterolytic cleavage of the O–O bond of H2O2 or ROOH assisted by two amino acid residues, a histidine and an arginine (Fig. 4) [80], which leads to a highly reactive iron(V)-oxo species. We then used as a hapten iron-α,α,α,β-meso-tetrakis(ortho-carboxyphenyl)porphyrin, α,α,α,β-Fe(ToCPP) (Fig. 2B). This hapten was chosen in the hope of generating such histidine or arginine amino acids, opposite to the ortho-carboxylate substituents of the phenyl rings. In addition, the metal-porphyrin was used, expecting that it could induce in the antibody an amino acid residue that could bind the metal and mimic the histidine proximal ligand of the iron atom in peroxidases.
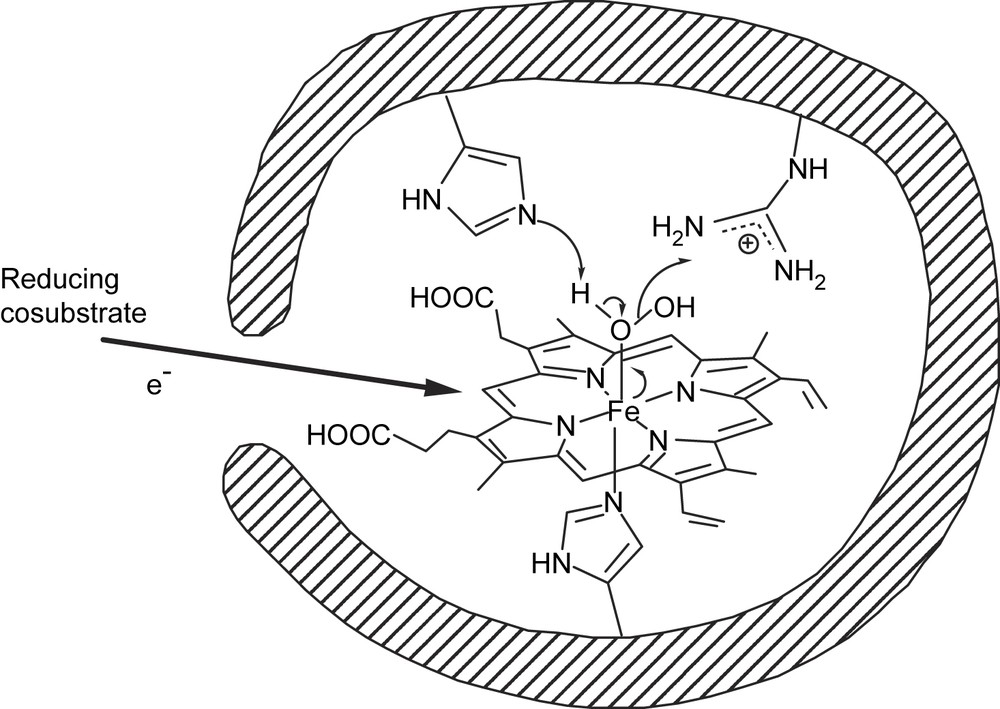
Schematic view of peroxidases and mechanism of the heterolytic cleavage of the O–O bond of hydroperoxides catalyzed by peroxidases.
Three monoclonal antibodies were found to recognize α,α,α,β-Fe(ToCPP). Two of them, 13G10 and 14H7, were able to bind it with Kd values of 2.9 × 10−9 M and 5.5 × 10−9 M, respectively. Those values are the best ones ever reported for the binding of an iron-porphyrin to an antibody [52,54–57,59,62,65]. UV–visible spectroscopy studies, associated with the determination by competitive ELISA of the apparent dissociation constants (Kd) for various porphyrins have allowed us to propose a possible binding site topology of antibodies 13G10 and 14H7 [70]. Both studies suggested first that the central metal atom of the hapten was not recognized by the antibody protein since α,α,α,β-Fe- and -Mn-ToCPP, as well as the free-base α,α,α,β-ToCPPH2, all bound to 13G10 with similar Kds [70]. This showed that no amino acid residue was induced in the antibody protein that was able to coordinate the iron atom of the hapten. Second, it appeared that the ortho-carboxylate substituents of the phenyl rings of the hapten were crucial for the recognition of Fe(ToCPP) by the antibodies, since the affinity of both 13G10 and 14H7 for this porphyrin was decreased by a factor of l02 and 103 when these substituents, were esterified (α,α,α,β-Fe[ToCMePP]) or shifted to the meta position (TmCPPH2). Finally, a comparison of the Kds for the four atropoisomers of ToCPPH2 (Fig. 2B) [70] showed that mainly three of the ortho-carboxylate substituents of the phenyl rings, in α,α,β positions, were recognized by amino acids of the antibody protein. This led us to propose a possible binding site topology in which roughly two thirds of the porphyrin macrocycle could be inserted in the binding pocket, with two adjacent α,α-carboxylates being more specifically bound to the protein. One phenyl bearing an α-carboxylate substituent could be outside the binding pocket, but the β-carboxylate, presumably the one bearing the linker to the carrier protein during immunization, could be on the edge of the pocket. These observations were recently confirmed by preliminary results of X-ray diffraction studies which led to the determination of the tridimensional structure of the Fe(ToCPP)–13G10 complex (Fig. 5) [81].
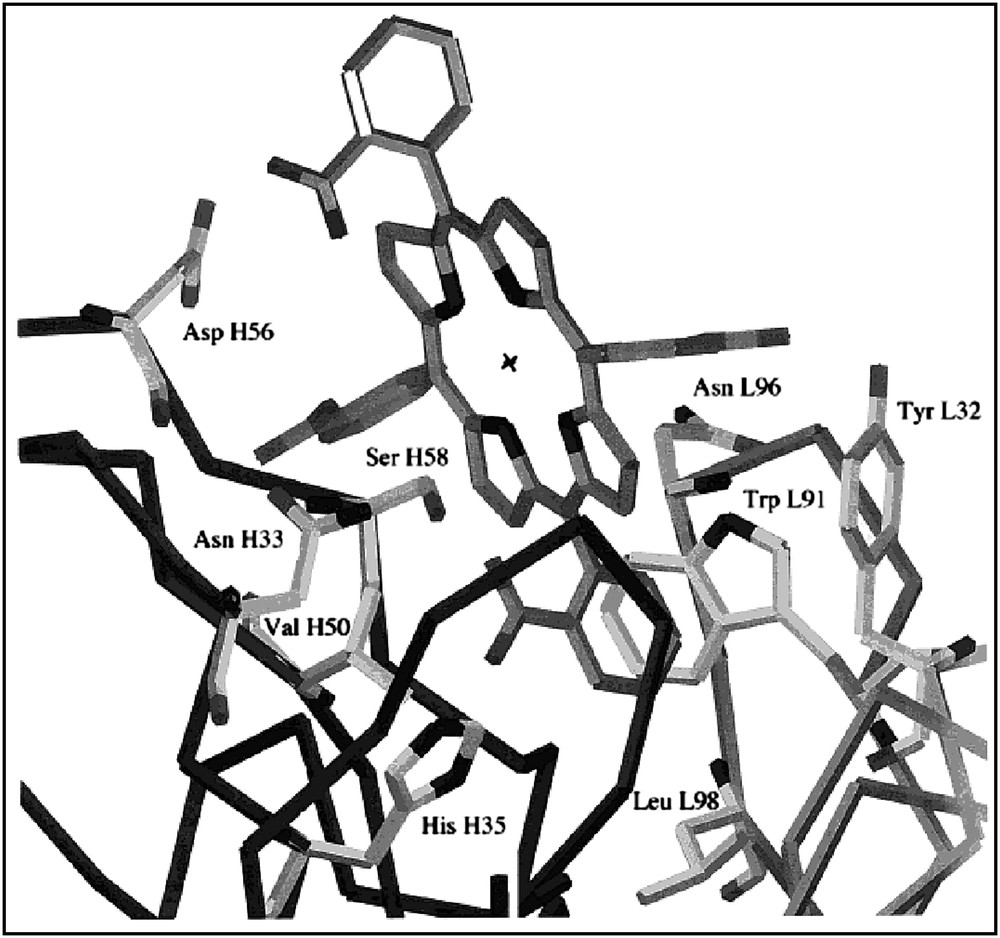
3D-structure of the 13G10–Fe(ToCPP) complex.
The two α,α,α,β-Fe(ToCPP)–13G10 and α,α,α,β-Fe(ToCPP)–14H7 complexes catalyzed the oxidation of 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) by H2O2 and it was clearly demonstrated [65] that the reaction did occur in the binding site of the antibodies. Both 13G10–porphyrin and 14H7–porphyrin complexes exhibited catalytic efficiencies about 5-fold higher than that obtained with free α,α,α,β-Fe(ToCPP) (Table 1). In this reaction, both the antibody proteins exerted a protecting effect towards the oxidative degradation of the porphyrin. Indeed, the rate of oxidation of ABTS by H2O2 remained constant for at least 1000 turnovers in the presence of both the antibody–Fe(ToCPP) complexes as catalysts, whereas in the presence of Fe(ToCPP) alone it slowed down after 15 min and the reaction stopped after only 40% conversion of ABTS. In addition, α,α,α,β-Fe(ToCPP)–13G10 was found able not only to catalyze the oxidation of a variety of co-substrates such as o-dianisidine, pyrogallol, ABTS and TMB (tetramethyl-benzidine) by H2O2, but also to use a wide range of hydroperoxides ROOH other than H2O2 as substrates, including alkyl-, aralkyl- and fatty acid-hydroperoxides [72].
Comparison of kinetic parameters for oxidation of various co-substrates by H2O2 catalyzed by iron-porphyrin–antibody complexes
| Complex [ref.] | Hapten | Substrate | kcat (min−1) | Km(H2O2) (mM) | kcat/Km (M−1 min−1) |
| HRP [64] | Leucomalachite green | 306 | 0.0005 | 6.1 × 108 | |
| FeIII-MPIXa–7G12 [51] | N-CH3-MPIX | o-Dianisidine | 394 | 24 | 1.6 × 104 |
| ABTSd | – | – | 1.4 × 104 | ||
| Pyrogallol | – | – | 7.3 × 103 | ||
| FeIII-MPIX [51] | o-Dianisidine | 166 | 43 | 3.8 × 103 | |
| FeIII-MPIX–9A5 [57] | N-CH3-MPIX | Pyrogallol | 132 | 35 | 3.7 × 103 |
| FeIII-MPIX–11D1 [57] | N-CH2OH-MPIX | Pyrogallol | 86 | 13 | 6.6 × 103 |
| FeIII-MPIX [57] | Pyrogallol | 21 | 100 | 2.4 × 102 | |
| FeIII-TpCPPb–13-1 L [58] | TpCPPH2 | Pyrogallol | 667 | 2.3 | 2.9 × 105 |
| FeIII-ToCPPc–13G10 [66] | Fe(ToCPP) | ABTS | 560 | 16 | 3.7 × 104 |
| FeIII-ToCPP–14H7 [61] | Fe(ToCPP) | ABTS | 63 | 9 | 7.1 × 103 |
| FeIII-ToCPP [61] | ABTS | 51 | 42 | 1.2 × 103 | |
| Fe-MPIX–2B4 [64] | N-CH3-MPIX | o-Dianisidine | 330 | 43 | 7.7 × 103 |
| Fe-MPIX [64] | o-Dianisidine | 77 | 25 | 3.1 × 103 | |
| MP8–3A3 [X] | MP8 | o-Dianisidine | 885 | 0.45 | 2 × 106 |
| MP8 [X] | o-Dianisidine | 590 | 0.4 | 1.45 × 106 |
a MPIX, mesoporphyrin IX.
b TpCPP, meso-tetrakis(para-carboxyphenyl)porphyrin.
c ToCPP, meso-tetrakis(ortho-carboxyphenyl)porphyrin.
d ABTS, 2,2-azinobis (3-ethylbenzothiazoline-6-sulfonic acid).
Studies of the kinetic parameters for the oxidation of ABTS by H2O2 at various pHs showed that the peroxidase activity of the α,α,α,β-Fe(ToCPP)–13G10 was optimal at pH 4.6, with a kcat value of 560 min−1 (Table 1), which led us to propose that a carboxylic acid side chain of the antibody could participate in the reaction mechanism. It could protonate one of the oxygen atoms of H2O2 and facilitate the release of a water molecule leading to the iron-oxo intermediate [72] (Fig. 6). Accordingly, the X-ray diffraction studies suggest that carboxylic acid side chain of an aspartate residue, Asp H56, could be responsible for this protonation step [81].
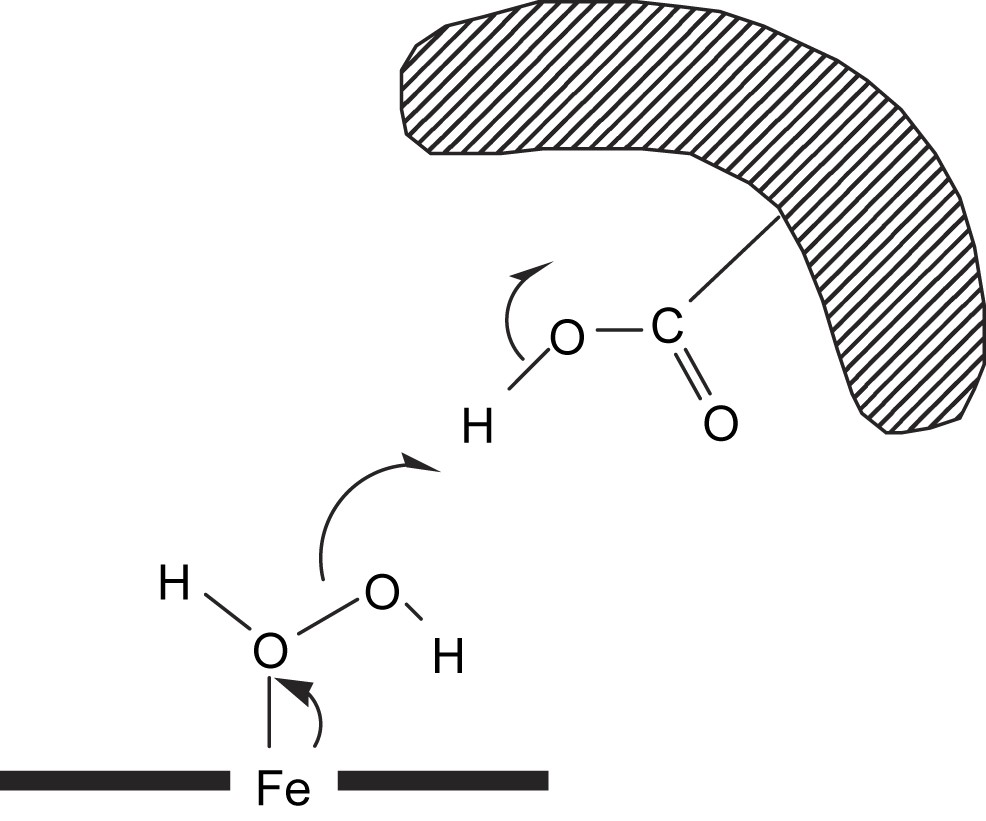
Possible mechanism of the heterolytic cleavage of the O–O bond of H2O2 catalyzed by Fe(ToCPP)–13G10 complexes.
3.1.1 Comparison of the kinetic parameters for the peroxidase reaction catalyzed by various antibody–Fe-porphyrin complexes
Antibodies have been elicited against meso-carboxyaryl substituted- [52,57,62,65–68,70,72,73], N-substituted [55,56,61,63,64,68,74], tin- [56,58] or palladium-porphyrins [59,60]. Several antibodies raised against N-substituted porphyrins [55,61,68] and meso-carboxyphenyl-substituted porphyrins [62,65] have shown, in the presence of their iron-porphyrin cofactor, a significant peroxidase activity.
The first monoclonal antibody, 7G12, was obtained by Cochran and Schultz [55] using N-methy1mesoporphyrin IX (N-CH3-MPIX) (Fig. 2A), as a hapten. The corresponding Fe(III)-MPIX–7G12 complex was shown to catalyze the oxidation of several typical peroxidase co-substrates, such as o-dianisidine, ABTS, and pyrogallol, by H2O2. The structure of a mesoporphyrin IX–7G12 complex was determined by X-ray diffraction studies [71]. It shows, like in the case of α,α,α,β-Fe(ToCPP)–13G10, that approximately 2/3 of the porphyrin are interacting with the antibody pocket, three pyrrole rings being packed tightly against residues of the VH domain and two pyrrole rings packed against tyrosine residues of the VL domain. In addition, a methionine H100c interacts specifically with one pyrrole ring and forces it to adopt a tilted conformation which should be favorable for the insertion of metal ions in the porphyrin ring and could explain the ferrochelatase activity of antibody 7G12.
The same N-methylmesoporphyrin IX hapten was also used by two other groups, [61,68] which, respectively, got two monoclonal antibodies 2B4 and 9A5. When associated with the Fe(III)-MPIX cofactor, those two antibodies were found to have a peroxidase activity: the 9A5–Fe(III)-MPIX complex was found to be able to catalyze the oxidation of pyrogallol by H2O2 [61] and the 2B4–Fe(III)-MPIX catalyzed that of o-dianisidine and ABTS, but neither that of pyrogallol nor that of hydroquinone by H2O2 [68].
After they got a first monoclonal antibody, 9A5, raised against N-methylmesoporphyrin IX, Feng et al. [61] also used N-hydroxymethy1mesoporphyrin IX (N-CH2OH-MPIX) (Fig. 2A) as a hapten, in which the oxygen atom of the hydroxymethyl group was supposed to mimic the oxygen atom of H2O2 when coordinated to the iron atom of heme in peroxidase. They obtained one monoclonal antibody, 11D1, which was able to catalyze, in the presence of Fe(III)-mesoporphyrin IX, the oxidation of pyrogallol by H2O2.
Finally, Takagi et al. [62] also used, like us, a meso-carboxyphenyl-substituted porphyrin as hapten, but this time the isomer bearing the carboxy substituent in para position on the meso-phenyl rings: meso-tetrakis(para-carboxyphenyl)porphyrin (TpCPPH2) (Fig. 2B). Not only did the corresponding Fe-TpCPP–antibody complexes catalyze the oxidation of ABTS and pyrogallol by H2O2, but the best catalyst for this reaction was found to be the complex associating Fe(TpCPP) with a recombinant antibody light (L) chain 13-1-L, which they named L-zyme.
The kinetic parameters for the oxidation of various co-substrates by H2O2, catalyzed by Fe-porphyrin–antibody complexes or by the corresponding free Fe(III)-porphyrins are compared in Table 1 with those reported for horseradish peroxidase [68,82,83].
In the case of Fe-porphyrin–antibody complexes, the kcat values range between 86 and 667 min−1, the best value being obtained for the oxidation of pyrogallol by H2O2 catalyzed by the Fe(TpCPP)–13-1-L complex [62]. Those values are 2.4- to 6.3-fold higher than those reported for iron(III)-porphyrins, which range from 21 to 166 min−1, which shows that iron-porphyrin–antibody complexes are better catalysts than the iron(III)-porphyrins alone. Iron(III)-porphyrin–antibody complexes also had a better affinity for H2O2 than free iron(III)-porphyrins, as shown by the Km values for H2O2, which range between 2.3 and 35 mM for Fe-porphyrin–IgG complexes, when compared to values of 43–100 mM for free Fe-porphyrins. As a consequence, iron(III)-porphyrin–antibody complexes exhibited higher efficiencies kcat/Km, ranging between 3.7 × 103 and 2.9 × 105 M−1 min−1, than the corresponding free iron(III)-porphyrins, with kcat/Km values ranging between 2.4 × 102 and 3.8 × 103 M−1 min−1. It is noteworthy that the best efficiency was obtained for the L-zyme, which exhibited both the higher kcat value, 667 min−1, and the lower Km value, 2.3 mM [62]. Unfortunately, those values cannot be compared to those corresponding to Fe(TpCPP) alone, which have not been reported. However, this kcat/Km value of about 2.9 × 105 M−1 s−1 remains far below those observed for peroxidases, which are among the most efficient enzymes, and exhibit kcat/Km values of about 6 × 108 M−1 min−1 [68,82,83]. This is mainly due to the very good affinity of the enzyme for H2O2, as shown by a Km value of about 0.5 μM.
3.2 Towards better mimics of hemoproteins: providing the iron atom of the antibody–porphyrin complexes with an axial ligand
As already mentioned above, the antibody–porphyrin complexes described so far are not as efficient catalysts as their natural hemoprotein counterparts. The ones which display a peroxidase-like-activity are characterized by kcat/Km values 4–3 orders of magnitude lower than those observed for natural peroxidases (Table 1), and the ones which display a monooxygenase-like activity do not catalyze very efficiently the oxidation of substrates such as alkenes. One reason for this failure could be the absence, in those antibodies, of an amino acid such as a histidine (peroxidases) or a cysteinate (cytochrome P450s) which axially coordinates the iron and enhances its redox potential [31,84]. To avoid this problem, two strategies were developed: (i) imidazole was added as a fifth ligand of the iron atom in 13G10–Fe(DoCPP) complexes [73], (ii) microperoxidase 8 (MP8), a heme octapeptide obtained by hydrolytic digestion of horse heart cytochrome c, was used as a hapten [85], since it contained in particular the imidazole of His 18 acting as the fifth axial ligand of the iron (Fig. 7).
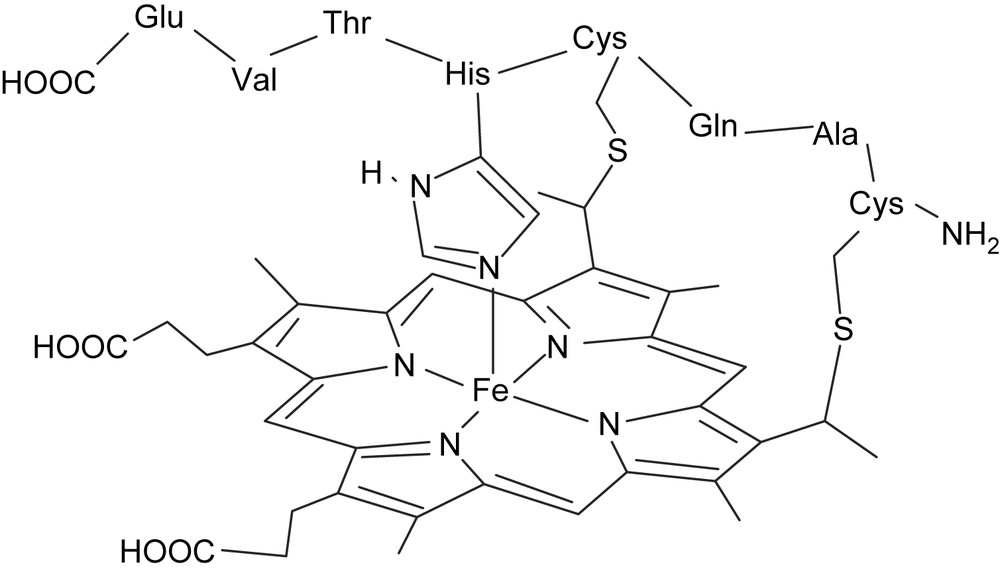
Schematic view of the structure of microperoxidase 8 (MP8).
3.2.1 Iron(III)-imidazole-porphyrin–antibody complexes
Since it was shown that none of the reported antibodies afforded any axial proximal ligand for the iron atom, the influence of imidazole on our antibody–Fe-porphyrin ligand complexes was investigated. All of the 13G10–Fe-porphyrin complexes only bound one imidazole ligand on the iron atom, but, whereas imidazole was found to inhibit the peroxidase activity of the α,α,α,β-Fe(ToCPP)–13G10 complex, it enhanced the peroxidase activity, by a factor 5–6, of the complexes of antibody 13G10 with less hindered iron(III)-α,α- and α,β-1,2-meso-di(ortho-carboxyphenyl)porphyrin, α,α- and α,β-1,2-Fe(DoCPP), which then constituted a nice model of peroxidase [73]. This could be explained by the fact that in the case of the more hindered Fe(ToCPP) porphyrin, imidazole was competing with H2O2 to bind to the iron on the less hindered face of the porphyrin, whereas in the case of the less hindered Fe(DoCPP) porphyrins, imidazole and H2O2 could bind simultaneously to the iron on the two opposite faces of the porphyrin, imidazole being able in this case to modulate the redox potential of the iron like the proximal histidine of peroxidases does.
3.2.2 Microperoxidase 8 antibody complexes
MP8 contains the heme prosthetic group together with the amino acid residues 14–21 of horse cytochrome c, and in particular His 18, whose imidazole group acts as the fifth axial ligand of the iron (Fig. 7). In addition, it also contains four carboxylate substituents, two from the propionate side chains of the heme, and two from the C-terminal glutamate (Glu 24) of the octapeptide, which could anchor it in the antibody binding site.
Finally, MP8 itself possesses a peroxidase-like and a monooxygenase-like activity. Indeed, at pH lower than 9, the sixth coordination position of the iron is occupied by a water molecule. This weak ligand can be replaced by H2O2, which allows the formation of highly oxidized intermediates, responsible for the two types of catalytic reactions. First of all, MP8 is able to perform the oxidation of several typical peroxidase co-substrates like o-dianisine [86], ABTS [87], guaiacol [88]. Second, MP8 also catalyzes the para-hydroxylation of aniline [89], the S-oxidation of sulfides [90], the monooxygenation of polycyclic aromatic compounds [91], and the N- and O-dealkylation of aromatic amines and ethers [92]. In addition, MP8 has also been recently shown to be able to oxidize N-monosubstituted hydroxylamines with formation of very stable iron(II)–nitrosoalkane complexes [93] and to catalyze the nitration of phenols by nitrite in the presence of H2O2 [94]. Unfortunately, MP8 presents two main drawbacks: first it dimerizes and aggregates at concentrations higher than 2 μM [85] and it is deactivated by oxidative degradation of the heme moiety [88]. Then, the two first objectives were, through the binding of MP8 to antibodies, to protect it against oxidative degradation of the heme moiety and to site-isolate it to keep it under a monomeric catalytically active form. The third objective was of course to induce a selectivity in the oxidation reactions catalyzed by MP8.
Among the seven monoclonal antibodies which recognized MP8, the one that had the best affinity for MP8, IgG1 3A3, bound it with an apparent Kd value of 10−7 M, which was in the range of the apparent Kd values already described for IgG–metalloporphyrin complexes [70,95].
UV–visible studies showed that the obtained 3A3–MP8 complex was an high-spin hexacoordinate iron(III) complex characterized by a spectrum very similar to that of MP8-FeIII with only a lower intensity of the Soret band at 396 nm. Such a phenomenon could be due to a simple hydrophobic interaction between MP8 and the antibody protein without any amino acid side chain acting as a sixth axial ligand of the iron.
The main characteristics of the binding site topology of antibody 3A3 could be determined from measurements, by competitive ELISA, of the apparent dissociation constants (Kd) for various heme derivatives. First, the central metal atom of the hapten was not recognized by the antibody since hemin, Mn-protoporphyrin IX and free-base protoporphyrin IX (PPIXH2), all bound to 3A3 with Kds of 3–5 × 10−7M. This confirmed the results of the UV–visible studies which indicated that no amino acid residue that was able to coordinate the Fe atom of the hapten was induced in the antibody binding site. Second, the octapeptide side chain of MP8 was not recognized by 3A3 since both hemin, which lacks this octapeptides, and MP8 bound 3A3 with an apparent Kd of about 10−7 M. Third, the carboxylate substituents of the hapten play a key role in the recognition of MP8 by the antibody. When these substituents are esterified or when these carboxylates are in ortho position on the meso-phenyl substituents of a tetraarylporphyrin, Kd is increased, respectively, by a factor of 103 and 2 × 102. In addition, to appreciate the size of the cavity left around the iron atom, the affinities of the tert-butyl-isonitrile ligand for the iron of MP8 or for that of 3A3–MP8, in the reduced and in the oxidized state, were compared. The corresponding ratios between the two Kd values, Kr were found, respectively, equal to 0.33 and 0.09. Such values, which are far below 1, are in favor of a steric hindrance brought by the antibody protein on the distal face of MP8 [96]. A possible active-site topology for antibody 3A3 was thus proposed (Fig. 8), in which MP8 binds to the antibody thanks to interactions of the carboxylates from the heme propionates and from the C-terminal glutamate of the octapeptide, at the opposite of the octapeptide chain, and the protein brings a partial steric hindrance of the distal face of MP8.
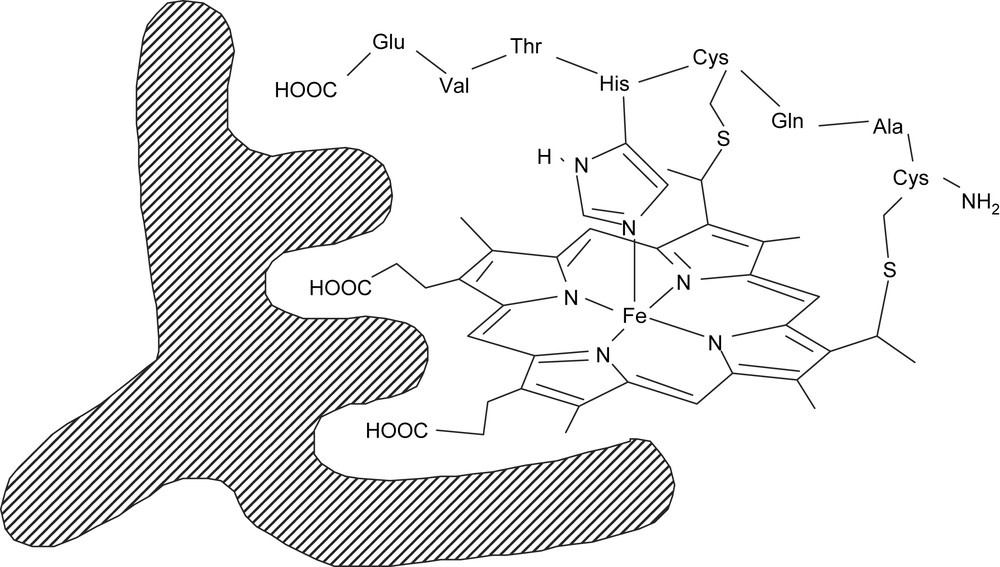
Possible binding site topology of anti-microperoxidase 8 antibodies.
The peroxidase activity of the 3A3–MP8 complex was analyzed and compared to that of MP8 alone. It appeared that 3A3–MP8 was a better catalyst than MP8 alone for the oxidation of four co-substrates: o-dianisidine, ABTS, guaiacol, and p-cresol [78]. These results are similar to those obtained by Kawamura-Konishi et al. [68] with the 2B4–MPIX-Fe complex as catalyst. For the four co-substrates, not only was the initial rate of oxidation slightly larger but also the percentage of co-substrate oxidized at the end of the reaction was 15–70% larger with 3A3–MP8, which indicates that the antibody has a protecting effect towards the oxidative degradations of MP8.
The kinetic parameters for the oxidation of o-dianisidine by H2O2 catalyzed by either 3A3–MP8 or MP8 were compared with those already reported in the literature for the oxidation of the various co-substrates by H2O2 catalyzed by porphyrin–antibody complexes (Table 1). The affinities of H2O2 for the MP8–antibody complex and for MP8 alone were very similar, as shown by the Km values observed for MP8 (0.4 mM) and for 3A3–MP8 (0.45 mM), and 3A3–MP8 exhibited slightly higher kcat (885 min−l) and kcat/Km values (2 × 106 M−l min−l) than MP8 alone (kcat = 590 min−l and kcat/Km = 1.45 × 106 M−l min−l). Those two observations confirmed that, on the distal face of MP8, there was no amino acid from the antibody participating to the catalysis of the heterolytic cleavage of the O–O bond of H2O2, which would have caused larger rate and efficiency enhancements. However, when compared to the values reported in the literature for other antibody–porphyrin complexes (Table 1), the kcat and kcat/Km values obtained in the case of 3A3–MP8 constitute the best ones ever observed for the oxidation of co-substrates by H2O2 catalyzed by Fe-porphyrin–antibody complexes. In particular, the kcat/Km values are 2–3 orders of magnitude higher than those observed for other Fe-porphyrin–antibody complexes, which arises both from a 100-fold better affinity of H2O2 for 3A3–MP8 than for other Fe-porphyrin–antibody complexes and from an about 2- to 3-fold higher kcat, due to the axial coordination of the proximal histidine His 18 on the iron atom of MP8.
3.3 Selective oxidations catalyzed by metalloporphyrin–antibody complexes
An early report from Nimri and keinan [75] described the first attempt to use this simple strategy to get models of cytochromes P450. Monoclonal antibodies were raised against Sn(IV)-meso-tetrakis (para-carboxyvinylphenyl)porphyrin (Sn-TpCVPP) (Fig. 2B). Five of those antibodies that bound the hapten with binding constants ranging between 2.5 × 10−8 M and 1.6 × 10−7 M were selected and the ability of the antibody–Mn(III)-TpCVPP complexes to catalyze the epoxidation of styrene by iodosylbenzene was then investigated. The best results were obtained with lyophilized antibody–Mn-TpCVPP complexes as catalysts, which produced turnover numbers ranging between 424 and 537 turnovers per 17 h of reaction, in CH2Cl2 at room temperature. Lyophilized Mn(III)-TpCVPP alone was not catalytic [56]. However, those turnover numbers were only 30–60% higher than that measured in the presence of a lyophilized mixture of a non-relevant Ab and Mn(III)-TpCVPP (300 turnovers/17 h). In addition, since no asymmetric induction was observed in any of these experiments, it seems unlikely that the reaction occurred inside the binding site of the antibodies, but more probably it was caused by Mn-porphyrin molecules bound unspecifically to the surface of the antibody protein.
A second short report by Liu et al. [74] described the production of monoclonal antibodies raised against N-4-bromophenyl-mesoporphyrin IX, the N-4-bromophenyl substituent being supposed to generate in the antibodies, binding site a hydrophobic pocket to accommodate aromatic substrates. Two of the obtained antibodies, 7C7 and 4D4, when associated with ferric mesoporphyrin IX, catalyzed the epoxidation of styrene by NaOCl with respective turnovers of 0.48 and 0.98 min−1, and the 4D4–Fe-mesoporphyrin IX catalyzed the N-demethylation of aminopyrine by H2O2 with a kcat of about 1 min−1 [74].
Finally, the most significant results were obtained recently by Nimri and Keinan [75] who raised monoclonal antibodies against a water-soluble tin(IV) porphyrin containing an axial α-naphthoxy ligand (Fig. 9). Such a stable complex was designed to mimic the postulated transition state of the reaction of S-oxidation of aromatic sulfides (Fig. 9). One of the selected monoclonal antibodies (SN 37.4) was found able, in the presence of a Ru(II) porphyrin cofactor, to catalyze the stereoselective sulfoxidation of aromatic sulfides such as thioanisole by iodosylbenzene, the S-enantiomer of thioanisole sulfoxide being obtained with a 43% enantiomeric excess (Fig. 9).
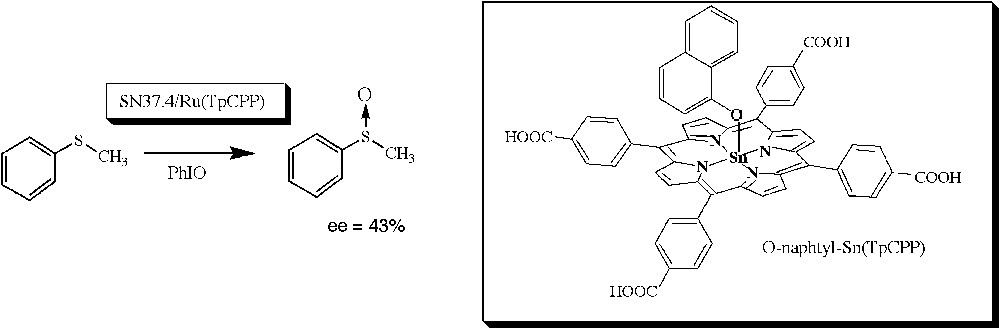
α-Naphthoxy–tin-porphyrin complex, used as a hapten for the generation of monoclonal antibodies able, in the presence of a Ru(II) porphyrin cofactor, to catalyze the stereoselective oxidation of sulfides [75].
We investigated the induction of stereoselectivity in oxidation reactions when using the 3A3–MP8 complex as a catalyst. Coordination chemistry was first used in order to appreciate the size of the cavity left around the iron atom in the binding site of the anti-MP8–antibody 3A3. This led us to show that the oxidation of N-monosubstituted hydroxylamines in the presence of the hemoabzyme 3A3–MP8-Fe(III) led to the formation of 3A3–MP8-Fe(II)–RNO complexes. This constituted a new reaction of hemoabzymes and the first example of fully characterized iron(II)–metabolite complexes of antibody–porphyrin complexes. In addition, the formation of such complexes appeared much more difficult with the bulky and hydrophobic N-(1-p-chlorophenylpropyl)hydroxylamine than with a rather small and hydrophilic hydroxylamine bearing a branched alkyl group, isopropylhydroxylamine. This confirmed that the antibody 3A3 brought a partial steric hindrance on the distal face of MP8, and, consequently, the influence of the antibody protein on two reactions catalyzed by MP8 was investigated.
First, it was shown that the 3A3–MP8 complex also catalyzed efficiently the nitration of phenol into 2- and 4-nitrophenol by NO2− in the presence of H2O2 (Fig. 10) [77].
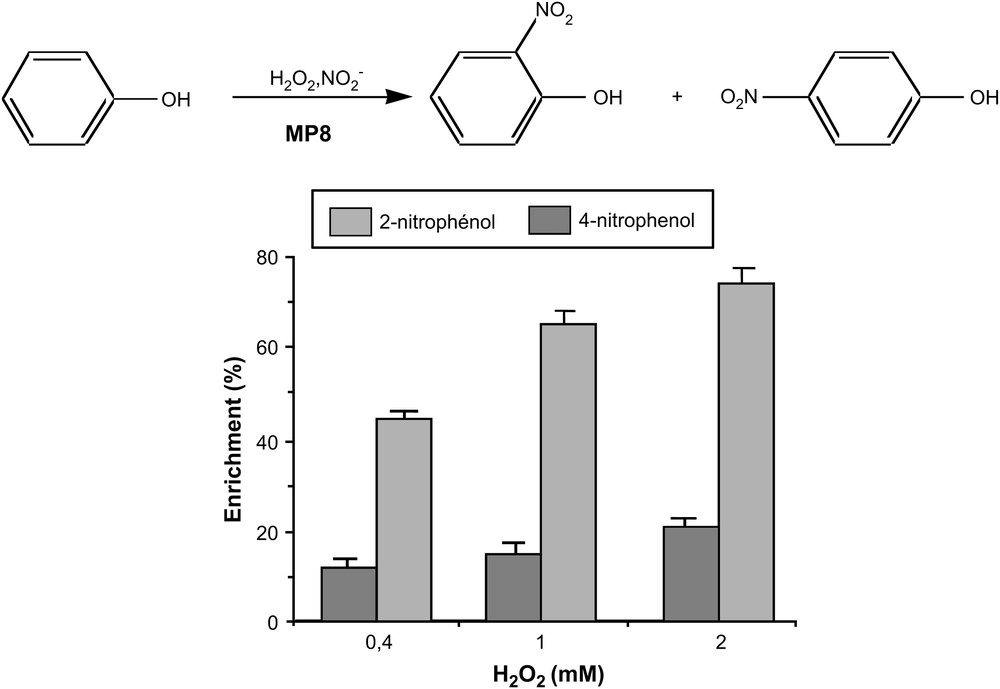
Nitration of phenol by H2O2/NO2− catalyzed by 3A3–MP8 [77].
pH dependance studies for this reaction confirmed that no amino acid from the antibody protein participated in the heterolytic cleavage of the O–O bond of H2O2. The inhibition of the reaction by cyanide and radical scavengers suggested an MP8-mediated peroxidase-like mechanism, involving the reduction of high-valent iron-oxo species by NO2− and phenol producing, respectively, NO2 and phenoxy radicals which then reacted to give nitrophenols. In those reactions also, the antibody protein appeared to have two major roles: it protected MP8 against towards oxidative degradations and it induced a regioselectivity of the reaction towards the formation of 2-nitrophenol.
Second, the influence of the antibody protein on the stereoselectivity of the S-oxidation of sulfides was examined [97]. Our results showed that MP8 alone and the antibody–MP8 complex were able to catalyze the oxidation of thioanisole by H2O2 and t-BuOOH, following a peroxidase-like “two-step oxygen-transfer mechanism” which involves a radical-cation intermediate. The best system associating H2O2 as catalyst, in the presence of 5% t-BuOH and 3A3–MP8 as catalyst, led to the stereoselective S-oxidation of thioanisole with a 45% enantiomeric excess in favor of the R isomer [97]. This constitutes the highest enantiomeric excess reported to date for the oxidation of sulfides catalyzed by porphyrin–antibody complexes and validates the use of the hemoabzyme 3A3–MP8 both as a catalyst for the selective oxidation of substrates and as a model system for hemoproteins. It is noteworthy that, on the contrary, a 43% enantiomeric excess in favor of the S-enantiomer of thioanisole sulfoxide was reported by Nimri and Keinan [75] with the SN 37.4–Ru(II) porphyrin complex as catalyst.
3.4 Comparison of the 13G10–Fe(ToCPP) and 3A3–MP8 complexes
The general features of the two antibody–Fe-porphyrin complexes, 13G10–Fe(ToCPP) and 3A3–MP8, are compared in Table 2.
General features of the two antibody–Fe-porphyrin complexes: 13G10–Fe(ToCPP) and 3A3–MP8
| 13G10–Fe(ToCPP) | 3A3–MP8 | ||
| Affinity for the iron-porphyrin | Kd = 10−9 M | Kd = 10−7 M | |
| Ligand of the iron | No | His | |
| Amino acid participating to catalysis | Asp? | No | |
| Protecting shield effect | Yes | Yes | |
| Control of the access to the iron | ? | Yes | |
| Catalysis | |||
| Peroxidase | kcat (min−1) | 560 | 885 |
| Km (mM) | 16 | 0.45 | |
| kcat/Km (M−1 min−1) | 3.5 × 104 | 2 × 106 | |
| Selective oxidations | No? | Nitrations | |
| Sulfoxidations |
It appears that both antibodies 13G10 and 3A3 share some common features. First of all, both of them have a good affinity for their porphyrin hapten, which is mainly due to the recognition of their carboxylate substituents. This particularly underlies the crucial role of carboxylates as strong determinants for the recognition of molecules by antibodies. Second, both antibodies exert a protecting effect towards the degradative oxidation of the iron-porphyrin cofactor, which is related, in the case of 13G10, with the insertion of the porphyrin moiety in a remarkably hydrophobic pocket. Third, in both antibody–porphyrin complexes, no amino acid side chain of the protein binds the iron atom, which confirms that the immunization of mice with a metal cofactor does not induce any recognition of the metal by the antibody. Finally both antibody–Fe-porphyrin complexes show a good peroxidase activity, with that of 3A3–MP8 being better, most probably because of the presence of the His 18 of the octapeptide chain, whose imidazole group acts as the fifth axial ligand of the iron.
Two main differences appear between the two antibody–Fe-porphyrin complexes. First, it seems that the carboxylic acid side chain of an aspartate participates in the catalysis of the O–O bond of H2O2 in the case of 13G10–Fe(ToCPP), whereas no amino acid from the antibody participates in catalysis in the case of 3A3–MP8. Second, in the latter case, the antibody brings a steric hindrance around the plane of the porphyrin and controls the access of substrates to the iron, leading to the selective oxidation of sulfides and nitration of aromatics, but most probably by a peroxidase mechanism. Such reactions were not observed in the case of 13G10–Fe(ToCPP).
As a conclusion, although they show an interesting oxidative activity, none of the two antibody–Fe-porphyrin complexes possesses all the element that are necessary for mimicking cytochrome P450-dependent monooxygenases. Consequently, other strategies, involving the covalent attachment of a metalloporphyrin cofactor to an anti-substrate antibody, were examined.
4 Semi-synthetic catalytic antibodies as models of cytochrome P450s: towards new strategies for elaborating artificial metalloproteins
This project aims at chemically modifying monoclonal antibodies in order to attach covalently an iron-porphyrin, to obtain artificial hemoproteins that would be able to catalyze the selective oxidation of substrates. In this new biocatalyst, the antibody will have the same role as the apoprotein in monooxygenases and will help in the selective positioning of the substrate with respect to the metalloporphyrin cofactor and will protect this cofactor towards oxidative degradations.
The general strategy presented in Fig. 11 has been derived from the one used by Pollack and Schultz [98,99] to modify antiphosphocholine Abs by covalent linkage of an arm ending in an imidazole, in order to enhance their ester-hydrolyzing activity. It involves four key steps as shown in Fig. 11: (1) the incubation of the anti-substrate Ab with the substrate modified by an arm including a disulfide bridge and ending with a reactive group such as aryl chloride, acid, or aldehyde; (2) the chemical reaction between a lysine residue of the Ab and the terminal reactive group of the modified substrate; (3) the reductive cleavage of the disulfide bridge by dithiothreitol (DTT), which leads to the Ab labeled with an arm ending with a free thiol group close to the binding site; and (4) the covalent coupling of the Fe-porphyrin to the thiol group. Such a strategy should lead to the positioning of the Fe-porphyrin close enough to the substrate binding site to catalyze its regioselective oxidation.
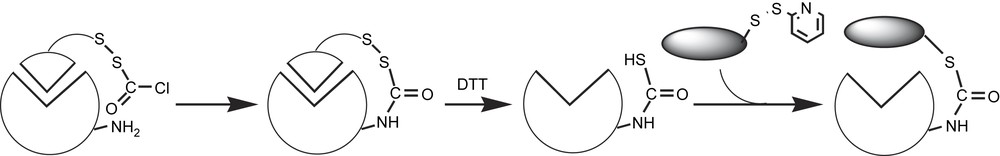
First strategy for the elaboration of semi-synthetic catalytic antibody with a peroxidase activity.
This strategy has been used to transform an anti-estradiol antibody into an artificial hemoprotein, with its Fe(III)-tetraarylporphyrin cofactor covalently linked to the protein, which could be able to catalyze, in the presence of a bulky imidazole, the regioselective oxidation of steroids by oxygen donors such as PhIO or H2O2.
The substrate chosen for the investigation was the testosterone, which has less affinity for the antibody (Kd = 1.7 × 10−7 M) than the estradiol (Kd = 9.5 × 10−10 M) has [100] and could be easily removed by extensive dialysis. In addition, its 3-keto function could be used as the starting point for the three-step synthesis of an arm, including a cleavable disulfide bridge and ending in a reactive bromoacetyl group (Fig. 12) [95]. Purified anti-estradiol antibodies were then incubated with the modified testosterone, and the covalent linkage of the arm was carried out under slightly basic conditions (Fig. 12). The disulfide bridge was further cleaved by reduction by DTT under nitrogen, and the testosterone moiety was eliminated by extensive dialysis under nitrogen (Fig. 12). Those two steps were controlled by competitive ELISA, using plates coated with the BSA–testosterone conjugate. Indeed, after the former step, anti-estradiol antibodies only retained 30% of affinity for the immobilized testosterone, compared to that observed for the starting unmodified antibodies. This was in agreement with the presence in, or close to, the binding pocket of a testosterone molecule covalently attached to the antibody protein, which prevented the binding of the antibody to the immobilized testosterone. Accordingly, after cleavage of the S–S bond by DTT and removal of the bound testosterone by dialysis, the affinity for the immobilized testosterone was almost totally restored, which showed that the covalently attached moiety did not affect the ability of the antibody to bind the testosterone–BSA conjugate.
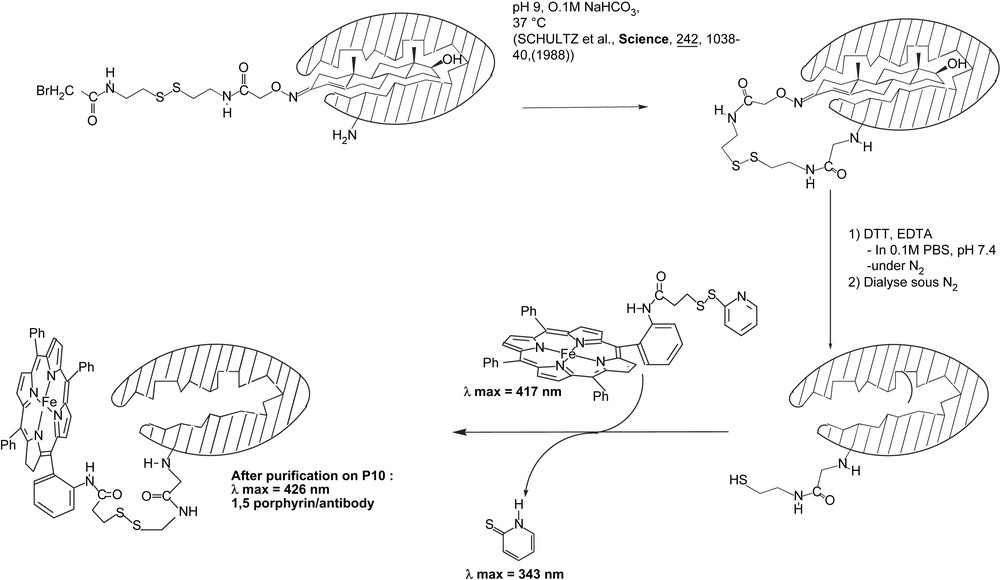
Covalent binding of mono-aminophenyl-tripentafluorophenylporphyrin on an anti-estradiol antibody.
In parallel, an Fe(III)-tetraarylporphyrin cofactor was synthesized by coupling meso-mono(ortho-aminophenyl)triphenylporphyrine (MAPTPPH2) (Fig. 12) [101,102] with N-succinimidyl-3-(2-pyridyldithiopropionate) (SPDP) (Fig. 12). This activated porphyrin was reacted with the previously prepared Ab (Fig. 12) and after purification on a P10 column, an Ab–porphyrin complex was isolated that exhibited a UV–visible spectrum typical of heme-proteins, with maxima at 280 and 426 nm (data not shown). Approximately 1.5 porphyrin was bound per antibody protein.
Unfortunately, after purification this new antibody–Fe-porphyrin complex revealed to be poorly soluble in water and did not show any significant oxidative activity.
A more straightforward strategy was envisioned to prepare those antibody-based artificial hemoproteins, which involves the synthesis of a new cleavable affinity label including the potential catalytic cofactor in four steps as described in Fig. 13: (a) the incubation of the antibody with the cleavable affinity label; (b) the chemical reaction between the side chain (A) of a targeted amino acid of the protein and the terminal reactive group of the compound (B); (c) cleavage of the labile linkage (disulfide bridge) which leads to the semi-synthetic antibody of interest; (d) release of the modified antigen by dialysis.
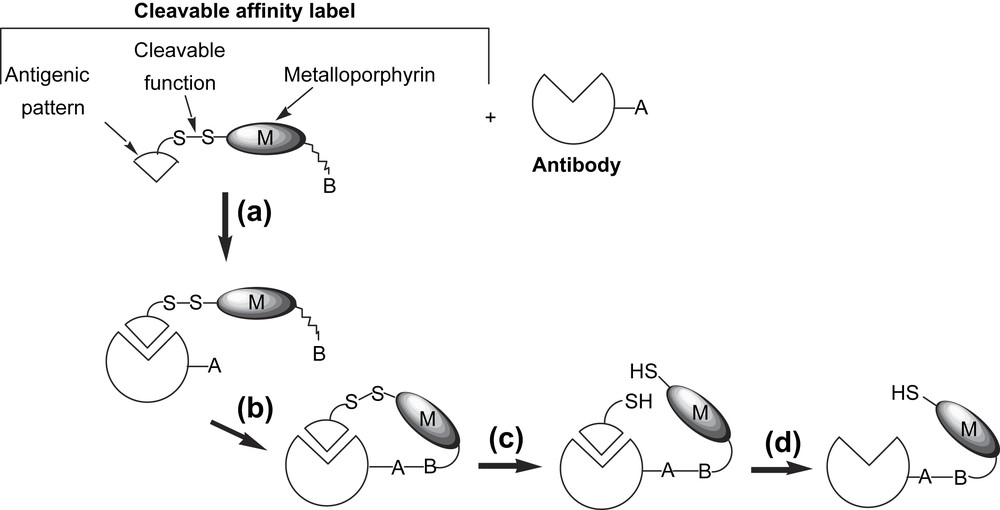
Strategy for the production of a semi-synthetic antibody able to catalyze the selective oxidation of steroids.
To fulfill the requirements of the strategy, we first synthesized the steroid–metalloporphyrin conjugate presented in Fig. 14A, which was designed to modify selectively an anti-estradiol antibody by chemical engineering (Fig. 14A). It included the following.
- – A testosterone moiety that would permit to direct selectively the synthetic cofactor in vicinity of the antibody binding site. This substrate was chosen, like in the first strategy, since it could be easily removed by extensive dialysis.
- – An iron complex of a meso-phenylsubstituted-porphyrin as catalytic cofactor which was chosen for several reasons. First, thanks to the aryl substituents in meso-positions, such porphyrins are protected from hydroxylations occurring at these positions, which lead to the macrocycle opening [103]. Second, meso-susbtituted porphyrins can be easily synthesized from the condensation of pyrrole with an arylaldehyde [104]. Third, metal complexes are efficient catalysts for the oxidation of substrates such as alkanes and alkenes [32,105]. Two consecutive meso-phenyls were substituted in ortho position, one by an arm including the cleavable disulfide bridge and ending in the testosterone moiety, and the other by a succinimidyl arm ending in a carboxylic acid group aimed at covalently binding the cofactor to the terminal amino group of a lysine residue of the antibody.
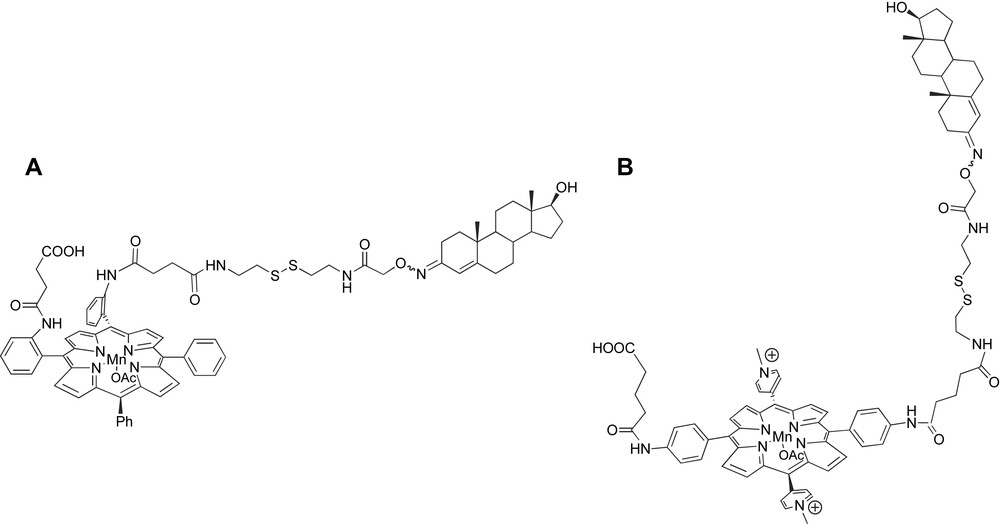
Mn(III)-porphyrin–steroid conjugates used as a new cleavable affinity label to get semi-synthetic catalytic antibodies.
Indeed, apart from the stability of the peptidic bond at pH 7.4 [106] compatible with the antibody, we had several reasons in favor of such a choice. First, the presence of available lysyl residues has been highlighted by a fluorimetric method [107] and by amino acid sequencing and 3D modelling of the variable regions of the antibody combining. Second, it was important to choose an amino acid with a reactive side chain of this kind, in order to optimize the selectivity of modification of the antibody. Finally, this type of residue has been already used by Pollack and co-authors for the obtention of semi-synthetic antibodies [98,99].
The porphyrin was synthesized by a strategy that involved a coupling reaction between a testosterone modified by an arm bearing a cleavable disulfide bridge and a meso-tetraarylporphyrin bearing two successive meso-ortho-amino-substituted-phenyl rings, α,α-5,10-bis-[{o-(2-carboxyethyl)carboxamido}phenyl]-15,20-diphenyl-porphyrin. The final porphyrin–steroid conjugate was successfully purified and fully characterized, and was subsequently metalated with manganese acetate. It was then coupled with the antibody using classical peptidic coupling reagents: N-hydroxysuccinimide and DCC. The obtained antibody–porphyrin-steroid was characterized by mass spectrometry (unpublished results). However, unfortunately, the yield of coupling was low, which we assumed to be due to the poor solubility in water of the steroid–metalloporphyrin conjugate. Consequently, the design of a new more water soluble porphyrin–steroid conjugate has been decided. In this conjugate, the central porphyrin ring bears not only an arm ending in a carboxylic acid for the selective coupling with the protein and an arm ending in the testosterone and including a cleavable disulfide bridge, but also two opposite cationic methyl-pyridinium substituents to increase its water solubility. The synthesis of the porphyrin–steroid conjugate aimed at building the modified Ab is still underway.
5 Conclusion
The production of new biocatalysts based on the association of monoclonal anti-porphyrin antibodies with metalloporphyrin cofactors has revealed to be a promising strategy for obtaining biomimetic systems of peroxidases. Indeed, two particular classes of antibodies raised against N-substituted [55,61,68] or meso-carboxyaryl-substituted [62,65,70] porphyrins have shown, in the presence of the corresponding iron(III)-porphyrin cofactor, a significant peroxidase activity characterized by efficiencies ranging between 3.7 × 103 and 2.9 × 105 M−1 min.−1. Those values are about 10-fold higher than those observed for the free Fe(III)-porphyrins, but they still remain 3–5 orders of magnitude lower than those of peroxidases, which are among the most efficient catalysts known, with kcat/Km values of 6 × 108 M−1 min−1 [68]. This shows that those biomimetic systems still remain imperfect, and, in particular, they present two main drawbacks. First, in most of the antibodies no amino acid residue such as histidine or arginine participating in the catalysis of the heterolytic cleavage of the O–O bond of peroxides has been generated. Indeed, the only residue which seems to participate in catalysis has been observed in the antibody 13G10 we have raised against Fe(ToCPP), in which a carboxylic side chain of an aspartic acid residue could protonate one oxygen atom of H2O2 and thus facilitate the release of a water molecule. Second, whatever the porphyrin hapten used, no axial ligand of the iron atom has been generated in the antibodies. However, the catalytic antibodies described up until now as models of peroxidases provide two advantages: they site-isolate the iron(III)-porphyrin, preventing its bimolecular oxidative self-degradation, and they control the diffusion of H2O2 to the porphyrin.
One way to improve those models and to find catalysts with better efficiencies consisted in providing the iron atom with an axial ligand which could have the same role as the distal histidine in peroxidases. This could be achieved either by adding an exogenous imidazole ligand to the iron to get iron(III)-imidazole-porphyrin–antibody complexes [73] or to raise antibodies against microperoxidase 8, a heme octapeptide containing a histidine bound to the iron atom [78]. Both strategies were successful as the association of imidazole with antibody–porphyrin complexes caused a 5- to 6-fold increase of the peroxidase activity and an antibody–microperoxidase 8 complex led to the best kcat/Km value ever reported for antibody–porphyrin complexes and 2 orders of magnitude only lower than that reported for horseradish peroxidase itself [73]. To get an even higher value, closer to that of the enzyme, would probably require the modification of the already existing antibodies, using site-directed mutagenesis, to provide the distal face of the porphyrin with two amino acids such as histidine and arginine that would improve the affinity for H2O2, thus decreasing Km, and participating in the catalysis of the heterolytic cleavage of the O–O bond.
It is not surprising that the strategy based on anti-porphyrin antibodies is particularly adapted to obtain models of peroxidases. Indeed, in those enzymes, the reducing co-substrate never enters their narrow active site, but only interacts with the meso edge of the heme, which is exposed to the solvent. This is probably what occurs in the antibody–metalloporphyrin complexes. The results of Green et al. [56], as well as the results on the study of the binding site of the 13G10 antibody [70,81], show that in those antibodies about two thirds of the porphyrin are included in the binding site leaving, one third of the porphyrin exposed to the solvent. On the contrary, in the case of cytochrome P450s, the substrate enters the active site and binds in a hydrophobic pocket, over the plane of the porphyrin close to the iron(V)-oxo species. Thus, the strategy based on anti-porphyrin antibodies seems less appropriate to obtain models of cytochrome P450s. Indeed, antibodies raised until now against rather flat porphyrins do not have any space over the plane of the porphyrin to bind the substrate [78,95], and it has not been possible to obtain antibodies having binding sites for both the substrate and the porphyrin, using as a hapten a porphyrin covalently linked to the substrate [95]. Then, although some positive results were obtained concerning the selective oxidation of substrates catalyzed by antibody–porphyrin complexes: (i) monoclonal antibodies raised against a water-soluble tin(IV) porphyrin containing an axial α-naphthoxy ligand were found able, in the presence of a Ru(II) porphyrin cofactor, to catalyze the stereoselective sulfoxidation of aromatic sulfides by iodosylbenzene [75]; (ii) antibody–microperoxidase 8 complexes have already proven to be able to catalyze the selective nitration of phenol derivatives [77] and the stereoselective sulfoxidation of aromatic sulfides by hydrogen peroxide [97], it is clear that some other strategies have to be envisioned.
The first one, that we have described above, involves the covalent attachment of a catalytic metal cofactor close to the recognition site of a monoclonal antibody, the role of which will be to position selectively the substrate with respect to the metal cofactor. This strategy is currently developed in our lab, for the covalent attachment of a metalloporphyrin close to the binding site of an anti-estradiol antibody, to obtain an artificial hemoprotein able to catalyze the regioselective and/or stereoselective oxidation of alkanes [95].
The second one, that we name the “Trojan Horse” strategy, is based on the high affinity of a protein for its substrate. In this strategy, the substrate, covalently linked to the catalytic metal cofactor, is used to anchor it non-covalently, but with a very high affinity, inside the binding site, the protein being responsible for the selectivity of the reaction catalyzed. This strategy has already been developed by Ward [108], who used the high affinity of avidin for biotin (Kd = 10−14 M) to elaborate new avidin–biotin–rhodium complex biocatalysts that were found able to catalyze the stereoselective reduction of acetamidoacrylic acid by H2. Such a strategy will be applied to the design of new biocatalysts for selective oxidations using the insertion of metalloporphyrin–biotin conjugates into avidin or streptavidin, and metalloporphyrin–steroid conjugates into anti-steroid antibodies.
Finally a third strategy, the so-called “host–guest” strategy can be envisioned. It involves the covalent or non-covalent insertion of a guest metal cofactor inside the cavity of a non-related host protein chosen not only to bring a chiral environment around the cofactor, but also, in some occasions, for its particular thermodynamic properties: thermoresistance, work under a wide range of pH. Such a strategy has been already used for the non-covalent insertion of a rhodium complex inside human serum albumin (HSA), to build up a hybrid metalloprotein that is able to catalyze the regioselective hydroformylation of styrene with CO and H2 [109]. We have thus chosen, to build up new hemoproteins, a simple strategy which involves the non-covalent incorporation of Fe(III)-α4-tetra-o-carboxyphenylporphyrin (Fe(ToCPP)), Fe(III)-tetra-p-carboxyphenylporphyrin (Fe(TpCPP)) and MP8 into thermostable β-1,4-endoxylanase or xylanase A (Xyl A) from Streptomyces lividans.