

1 Introduction
Dopamine β-monooxygenase (DβM) is a copper-containing monooxygenase which catalyzes the benzylic hydroxylation of dopamine (DM) into norepinephrine (NE, Scheme 1a), two important neurotransmitters involved in the central and peripheral nervous system of mammals [1]. DβM, which exists in soluble and membrane-bound forms, can be isolated from bovine medullae in its soluble form [2]. The active enzyme is a 290-kDa tetramer that incorporates two copper ions per subunit for optimum activity [3]. The copper ions are weakly bound to the protein [4] and cycle between +I and +II oxidation states during catalysis, as observed by EPR measurements [5]. In addition to copper, DβM requires two other cofactors for activity, reducer ascorbate and dioxygen.
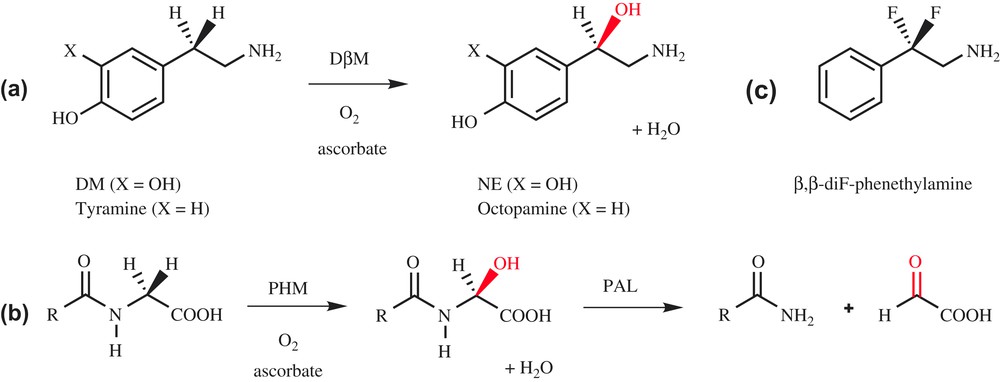
Hydroxylation catalyzed by DβM (a) and PHM (b).
DβM shares high similarities with the peptidylglycine α-hydroxylating monooxygenase (PHM) domain of peptidylglycine α-amidating monooxygenase (PAM). PAM catalyzes the biosynthesis of peptidic hormones, which contain an amide linkage at their C-terminus (substance P, oxytocin, thyrotropin and calcitonin). To perform this task, PAM possesses two different domains, a peptidylglycine α-hydroxylating monooxygenase (PHM) domain, corresponding to a monooxygenase activity, and a peptidyl-α-hydroxylglycine α-amidating lyase (PAL) domain, corresponding to a lyase activity (Scheme 1b) [6]. PHM and DβM require the same cofactors [7], and they possess sequence homologies in their catalytic domain [8]. Moreover, the copper ions in both enzymes undergo redox cycling [9] and have similar coordination spheres. While DβM has been the subject of extensive structural investigations, there is no crystallographic data available to date on this enzyme. Nevertheless, structural information can be deduced from the X-ray structure of PHM, which was recently described (Fig. 1a) [10]. This structure confirms that PHM contains two type-II copper atoms per active site, CuH, which is coordinated to three histidine residues and a water molecule, and CuM, bound to two histidine and one methionine. CuM is the binding site for co-crystallized substrate. An important feature revealed by the X-ray structure is that CuH and CuM are located in two different domains separated by a solvent pocket, and are distant from 11 Å. This distance suggests that copper ions are quite far apart in DβM as well, as already deduced from magnetic coupling measurements [4].

X-ray structure of oxidized PHM: (b) structure of the binuclear active site of PHM [10], (a) structure of the O2 binding site [18]. O2 (red) is shown bound to CuM (green) in an end-on manner. (For interpretation of the references to color in figure legends, the reader is referred to the web version of this article.)
Many studies have been devoted to the understanding of the mechanism of DβM. During catalysis, both copper ions are reduced to CuI, but interaction with the substrate and oxygen active species is believed to occur only at CuM, as suggested by the structure of PHM co-crystallized with its substrate [10]. Dioxygen activation and O-atom transfer catalyzed by DβM thus involve two copper ions, but take place in a mononuclear-like mechanism. Regarding the mechanism of C–H activation, structure–reactivity correlations on DβM with a series of ring-substituted phenethylamines which exhibited a slope at a significantly faster rate with electron-donating substituents than with electron-withdrawing substituents were initially interpreted in terms of an electron-deficient transition state resulting from H-atom transfer between substrate and activated O2 [11]. However, the H-transfer reaction has been reformulated in the context of a modified Marcus model, whereby hydrogen transfer occurs quantum mechanically and trends in reactivity are expected to reflect the impact of the reaction driving force on the rate [12]. Regarding the nature of the various copper–oxygen species involved in O2 activation (Scheme 2), an extensive and open debate has taken place in the recent literature [13].
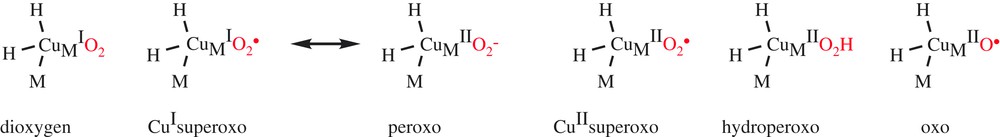
Candidates for the copper–oxygen species in DβM and PHM.
Mechanisms previously proposed for DβM involved the accumulation of an activated oxygen intermediate such as a hydroperoxo (Scheme 3, path b) or oxo species (Scheme 3, path c) before substrate activation. Regarded for a long time as the copper–oxygen active species that is responsible for the H-atom abstraction, the hydroperoxo species is, however, not a good candidate for the activated O2 species in DβM and PHM, since experimental data indicate a decreasing magnitude for the O-18 kinetic isotope effect as the rate constant for C–H activation fell over 2.5 orders of magnitude [12].
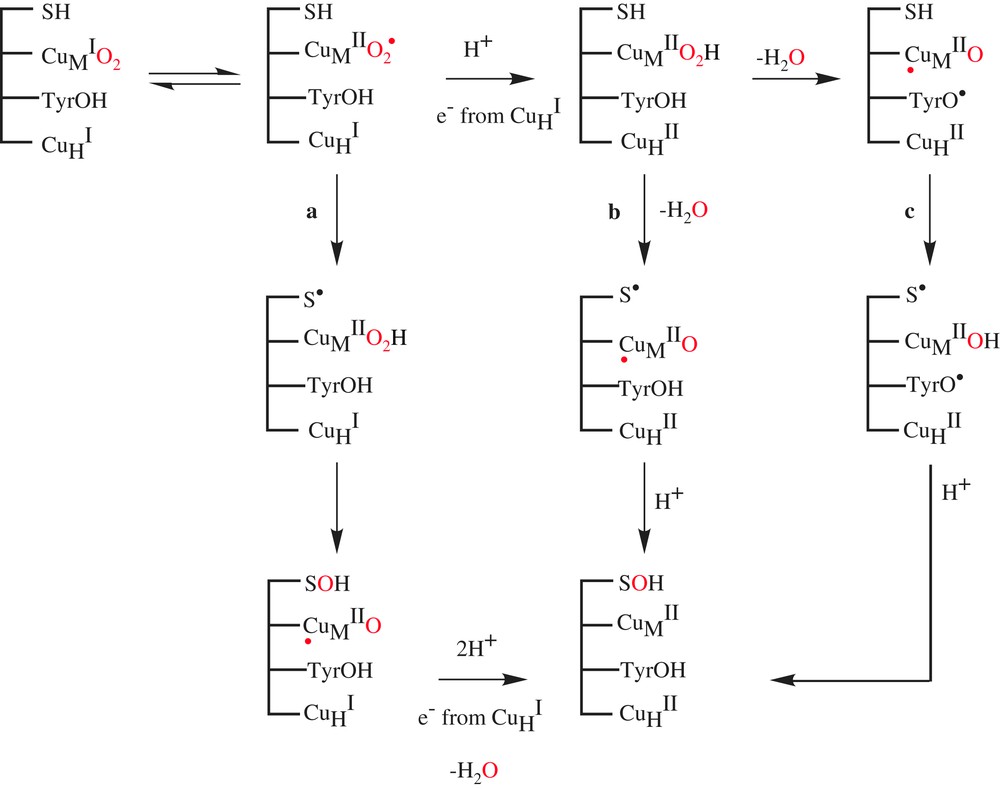
Mechanisms for the hydroxylation catalyzed by DβM and PHM.
Formation of the highly reduced metal-oxo was proposed to occur via the reductive cleavage of the hydroperoxo species by a conserved active site tyrosine (Scheme 3, path c) [14]. While some available experimental data on DBM and PHM could be in favor of the occurrence of tyrosine residue in activation of CuMIIO2H [15], the preparation of the tyrosine mutant of PHM (Y318F) indicated no impact on kcat and, most significantly, less than a 4-fold reduction in the rate of C–H activation. This site-specific mutagenesis experiment effectively eliminated tyrosine residue in activation of CuMIIO2H from consideration [16].
Recently, Klinman et al. reported two experimental probes of the activated oxygen species in DβM [17]. They have synthesized the substrate analog β,β-difluorophenethylamine (Scheme 1c) and examined its capacity to induce reoxidation of the pre-reduced copper sites of DβM upon mixing with O2 under rapid freeze-quench conditions. This experiment failed to give rise to an EPR-detectable copper species, in contrast to a substrate with a C–H active bond. This indicates either that the reoxidation of the enzyme-bound copper sites in the presence of O2 is tightly linked to C–H activation or that a diamagnetic species CuMIIO2 has been formed. Klinman et al. conclude that there is no accumulation of an activated form of O2 before C–H abstraction in DβM, presenting a mechanism in which a diamagnetic superoxo complex CuMIIO2, formed initially at very low levels, abstracts a H-atom from the substrate to generate CuMIIO2H and a substrate-free radical as intermediates. Subsequent participation of the second copper site per subunit completes the reaction cycle, generating the hydroxylated product and water (Scheme 3a).
Focusing on PHM, Prigge et al. [18] succeeded in trapping a copper–dioxygen complex by freezing protein crystals that had been soaked with a slow substrate and ascorbate in the presence of O2. The X-ray crystal structure of this pre-catalytic complex, determined to 1.85-Å resolution, reveals that oxygen binds to one of the coppers in the enzyme with an end-on geometry (Fig. 1b). This result could confirm Klinman's proposal; however, the structure gives no indication if the copper–dioxygen complex is a CuMIO2 species or a CuMIIO2 species and if these species are responsible for the H-atom abstraction of the substrate.
More recently, mechanisms of dopamine hydroxylation by the CuIIO2 species and CuIIO species of DβM was investigated using QM/MM calculations for a whole-enzyme model of 4700 atoms [19]. A calculated activation barrier for the H-atom abstraction by the CuIIO2 species is 23.1 kcal/mol, while that of the CuIIO is 5.4 kcal/mol. Energies of the optimized radical intermediate in the superoxo- and oxo-mediated pathways are 18.4 and −14.2 kcal/mol, relative to the corresponding reactant complexes, respectively. These results demonstrate that the CuIIO species can better mediate dopamine hydroxylation in the protein environment of DβM.
In order to better understand the nature of copper–oxygen species and the mechanism of O2 activation catalyzed by this class of enzymes, we studied the behavior of DβM with superoxide anion as oxygen atom donor. In an earlier paper, using a non-biological system for the production of superoxide (KO2, DMSO and 18-crown-6), Henry et al. [20] established that superoxide anion could be a cofactor for DβM. We thus decided to focus our study on the role of superoxide generated by a biological system, continuously released in the neighborhood of DβM. In this paper, we describe a DβM activity with superoxide generated by the xanthine/xanthine oxidase (XOD) system. We also show that in the absence of its substrate, DβM possesses a SOD activity. We determine the influence of the concentration of each species necessary for activity, evidence a pH-dependence, and propose a mechanism for superoxide-assisted DβM activity.
2 Experimental procedures
2.1 Materials
Xanthine, tyramine hydrochloride, bovine liver catalase, bovine erythrocytes superoxide dismutase and cytochrome c (Cyt. c) were purchased from Sigma. Xanthine oxidase from cow milk was purchased from Roche Diagnostics (Meylan, France).
2.2 Enzyme purification
Bovine adrenal glands were collected from the slaughterhouse. Medullae were immediately isolated and stored at −80 °C. DβM was later purified according to a procedure previously described by Ljones [21], except that the ConA-Sepharose affinity column was replaced with a Phenyl-Sepharose column. Purified DβM was more than 90% pure, as judged by SDS-PAGE (see Fig. 1 in Supplementary material), and exhibited a specific activity of 16–20 units/mg. Protein contents were measured using the Bradford assay from Bio-Rad and BSA as a standard.
2.3 SOD activity measurement
SOD activities were measured according to the classical Cyt. c assay [22]. All measures were performed in 50 mM phosphate buffer at pH 7.8.
2.4 DβM activity assay
Tyramine hydrochloride was used as a substrate for DβM and production of octopamine was monitored by HPLC. DβM was mixed with XOD, xanthine and tyramine for activity. Samples containing xanthine (1.4 mM), tyramine (10 mM), variable amounts of XOD (0–2.5 mU), DβM from 32 to 160 μg/mL, and 50 mM phosphate buffer, and had a final volume of 50 μL. Unless when mentioned, samples also contained bovine liver catalase at 1000 U/mL. Activity measurements at pH 6.1 and 6.4 were performed after introduction of acetic acid to final concentrations of 10 mM and 5 mM, respectively. Samples were left to stirring for 30 min at 25 °C, and stopped by addition of orthophosphoric acid (4 μL). Samples were next completed to 200 μL by 50 mM phosphate buffer at pH 7.2.
Resulting mixtures were analyzed using HPLC. Samples were injected on a reverse-phase C18 column using a HP Series 1100 apparatus. The eluting solution was a mixture of 5 mM orthophosphoric acid and acetonitrile. The initial rate of acetonitrile of 0.5% was maintained for 6 min, and it was increased linearly to reach 20% after 6 more minutes, under a constant flow of 1 mL/min. Analysis of outgoing species was performed by following the absorbance at 240 nm. Retention times in such conditions were octopamine, 4 min; uric acid, 5.8 min; xanthine, 7 min; and tyramine, 10 min.
3 Results
3.1 DβM SOD-like activity
We first investigated the SOD-like activity of DβM. A SOD activity measurement was performed with DβM, using the Cyt. c assay monitored by UV–vis spectroscopy at 25 °C [22]. Purified bovine DβM added to a solution containing Cyt. c and the XOD system at pH 7.8 showed a specific superoxide dismutase activity of 41 ± 5 U/mg that was earlier found as 10 U/mg by Henry et al. [20]. When comparing the activity of a DβM monomer to that of a bovine erythrocyte CuZn–SOD monomer, that is the activity of a copper center for each enzyme, DβM dismutase activity is about 3.7% of the latter. DβM thus presents a very low reactivity towards superoxide anion.
3.2 Time-dependent substrate hydroxylation catalyzed by XOD-assisted DβM
We determined whether addition of substrate tyramine to DβM in the presence of XOD-generated superoxide could yield hydroxylated product octopamine. We thus mixed the XOD system with tyramine and DβM, in the absence of any other cofactor, and we observed that the production of octopamine was time-dependent over a time period of 30 min. The resulting activity was 4.5 nmol/min/mg at pH 7.6 and 25 °C. Uric acid formation was also quantified, and in our conditions, octopamine formation parallels that of uric acid, hence also superoxide anion production (Fig. 2). Indeed, transformation of 1 mol of xanthine to uric acid by XOD yields about 0.25 mol of superoxide ion at pH 7.6 [23].
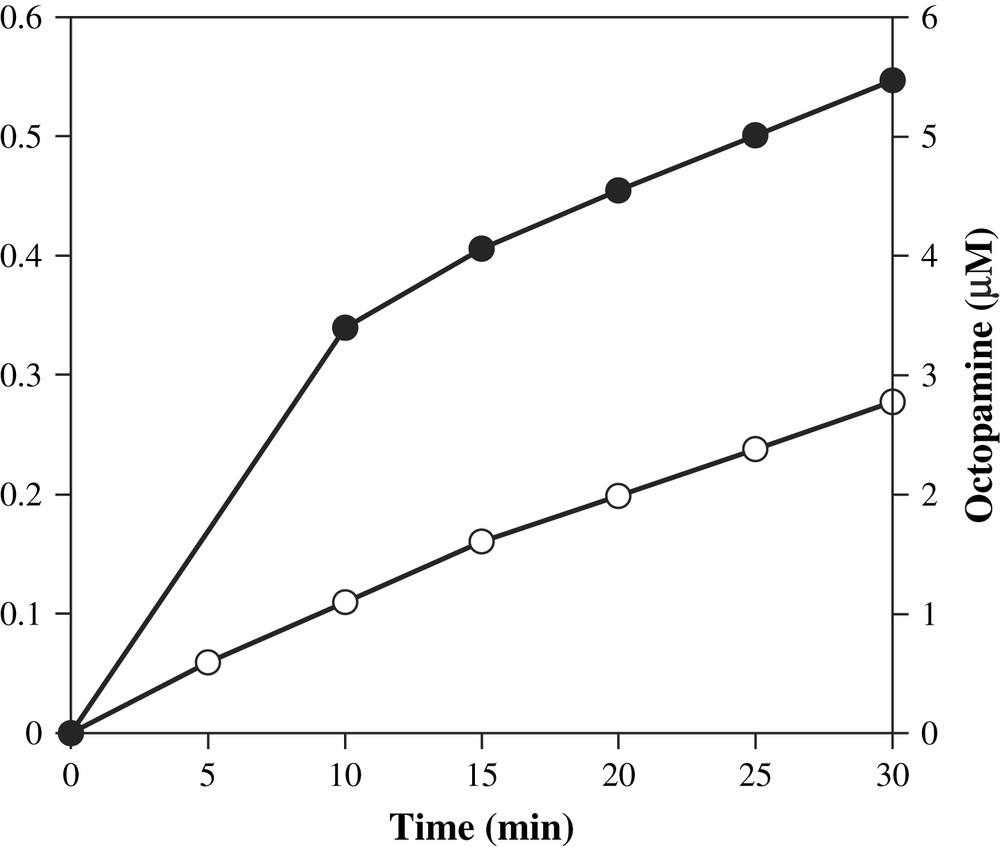
Time-dependent octopamine (●) and uric acid (○) formations at 25 °C. Aliquots were removed from a mixture containing XOD (0.9 mU), xanthine (1.4 mM), tyramine (10 mM), DβM (40 μg/mL), catalase, phosphate buffer (50 mM), at a final pH of 7.6, and analyzed for activity by HPLC.
In order to determine which species was responsible for the activation of DβM, we modified our system and performed various blank experiments. The involvement of DβM in octopamine formation was first confirmed. Absence of bovine DβM definitely prevents the formation of octopamine, while uric acid production remains unchanged (Fig. 3, −DβM). Since DβM SOD-like activity and XOD system generate H2O2 [24], which is a known DβM inhibitor [25] and might be responsible for undesired by-reactions, catalase was added to activity buffer. Addition of catalase to the reference mixture simultaneously produced a 120% increase in octopamine and uric acid production (Fig. 3, +Cat). The role of XOD was next confirmed. With no XOD added, i.e. after incubation of DβM with xanthine and tyramine in phosphate buffer at pH 7.6, no uric acid and little octopamine formation could be detected (Fig. 3, −XOD).
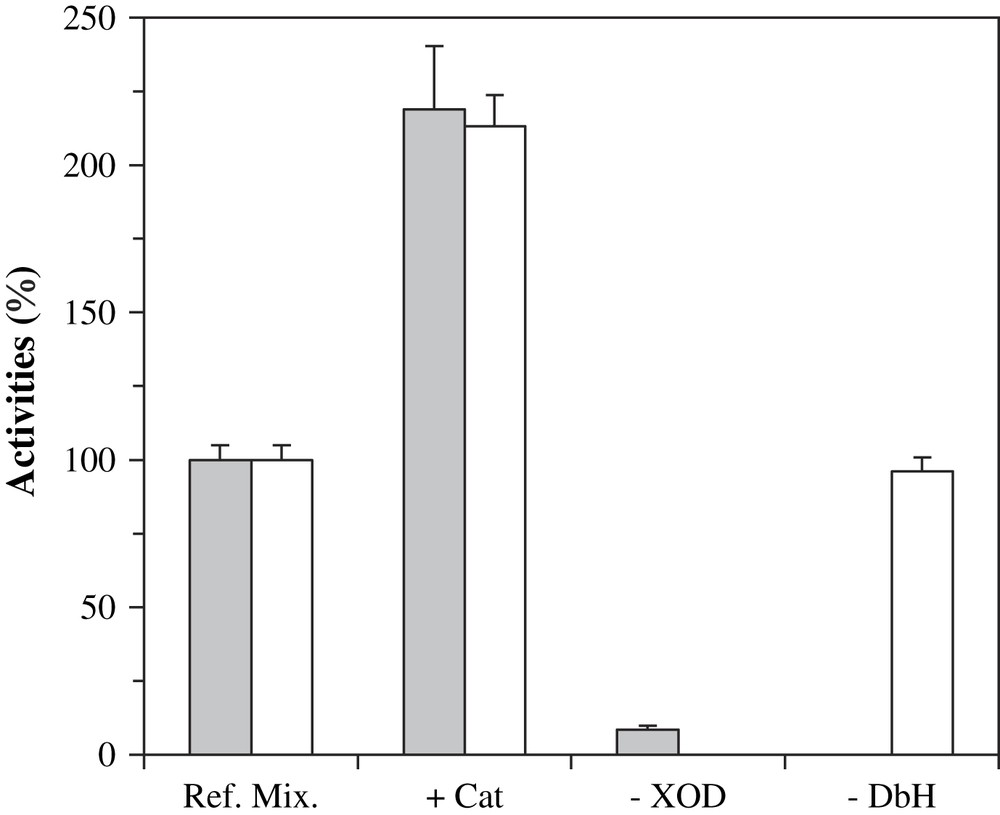
Octopamine (gray bars) and uric acid (white bars) formation in incomplete mixtures after 30 min at 25 °C. Reference mixtures contained XOD, 1 mU, xanthine, tyramine and DβM in phosphate buffer at pH 7.6. +Cat: ref. mix. with catalase added at 1000 U/mL. −XOD: ref. mix. without XOD. −DβM: ref. mix. deprived of DβM. In the following figures, conditions of ‘ref. mix. +Cat’ will be coined as ‘standard assay’.
A last experiment was performed in order to confirm and definitely prove the involvement of superoxide anion in the activation of DβM by the XOD system. Addition of catalase to the XOD system allows only superoxide to be produced concomitantly with uric acid. Selective removal of O2− would unambiguously answer this question. Thus, to a solution containing xanthine, XOD, tyramine, DβM and catalase, various amounts of CuZn–SOD were added, and the octopamine formed after 30 min was quantified (Fig. 4). Addition of 0.5 SOD unit was responsible for a 62% decrease in DβM activity as opposed to the complete system deprived of SOD. With 25 units added, the remaining activity was about 10%, a value which is close to the background activity obtained in the absence of XOD (Fig. 4). This experiment proves that superoxide anion is the molecule that is involved in octopamine formation assisted by the XOD system.
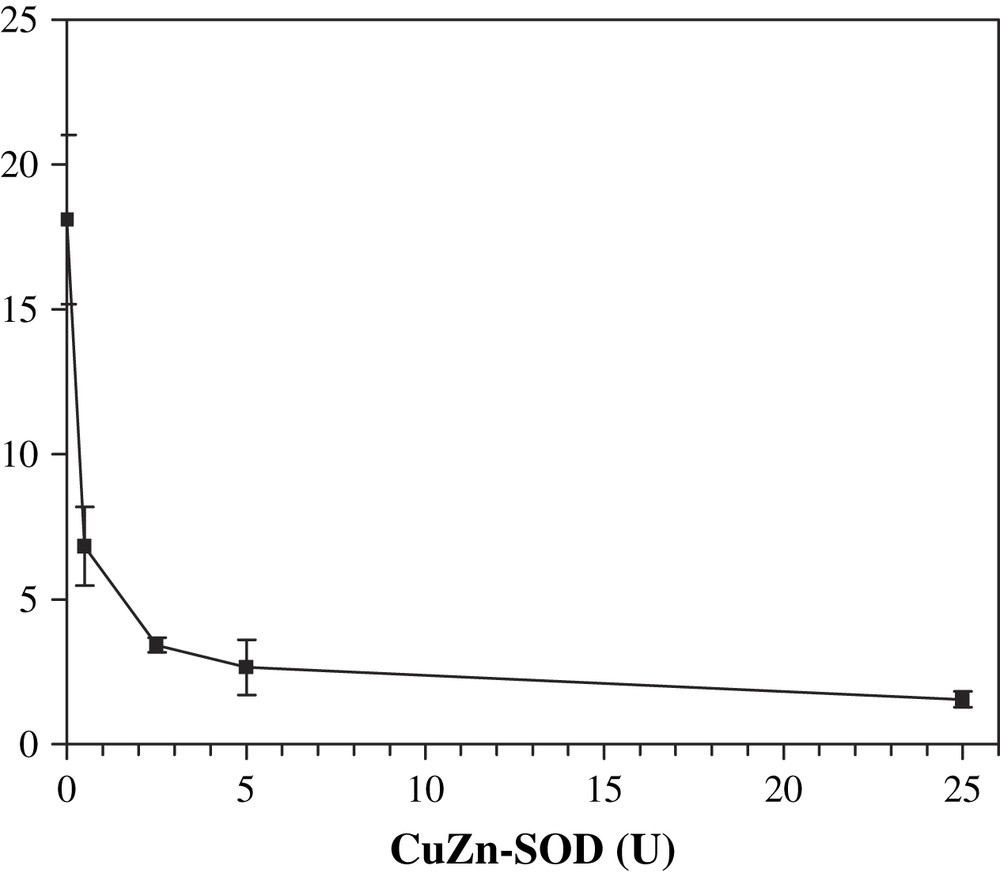
Octopamine formation after 30 min at 25 °C in the presence of SOD. Solutions contained XOD, xanthine, tyramine, DβM, 80 μg/mL, catalase and SOD from 0.5 to 25 units. Octopamine concentration (μM) was determined by HPLC. Each point is the average of three different experiments ± standard deviation.
3.3 pH study
The physiological pH for DβM is about 5.5, and most in vitro experiments are performed at a pH near 6.0. The activity of XOD is also pH-dependent, with an optimum activity around pH 8.0. Therefore we wished to determine how octopamine production by the XOD/DβM combined system was modified by an alcalinization of the medium. In order not to have a too low activity for the XOD system, our study was initiated at pH 6.1 in phosphate plus acetate buffer, up to a pH of 8.2. The pH study was restricted to this pH range so that the influence of the unchanged buffer could be neglected and a modification in activity due to the presence of a buffer ion could be avoided. At pH 6.1, octopamine formation could be evidenced, in spite of the low XOD activity. Increasing pH up to 8.2 led to an increasing octopamine formation, with similar activities at pH 7.6 and 8.2 (Fig. 5). The production of uric acid was also followed. From these data, the ratio between octopamine and uric acid concentrations after 30 min incubation, i.e. the pure DβM activity, was extracted. This activity was found maximal at pH 6.8 with about five times the activity observed at pH 6.1, and 1.2 times the activity at pH 8.2. Thus the use of the XOD system as a cofactor for DβM shows an optimum activity at a pH more than 1 unit superior to its physiological pH. Moreover, the decrease in activity at values superior to neutral pH is slow, octopamine to uric acid ratios being similar at pH 7.6 and 8.2. This system allows an important shift in the pH for optimum activity, thus showing the stability of DβM with regards to alcalinization, as well as its versatility.
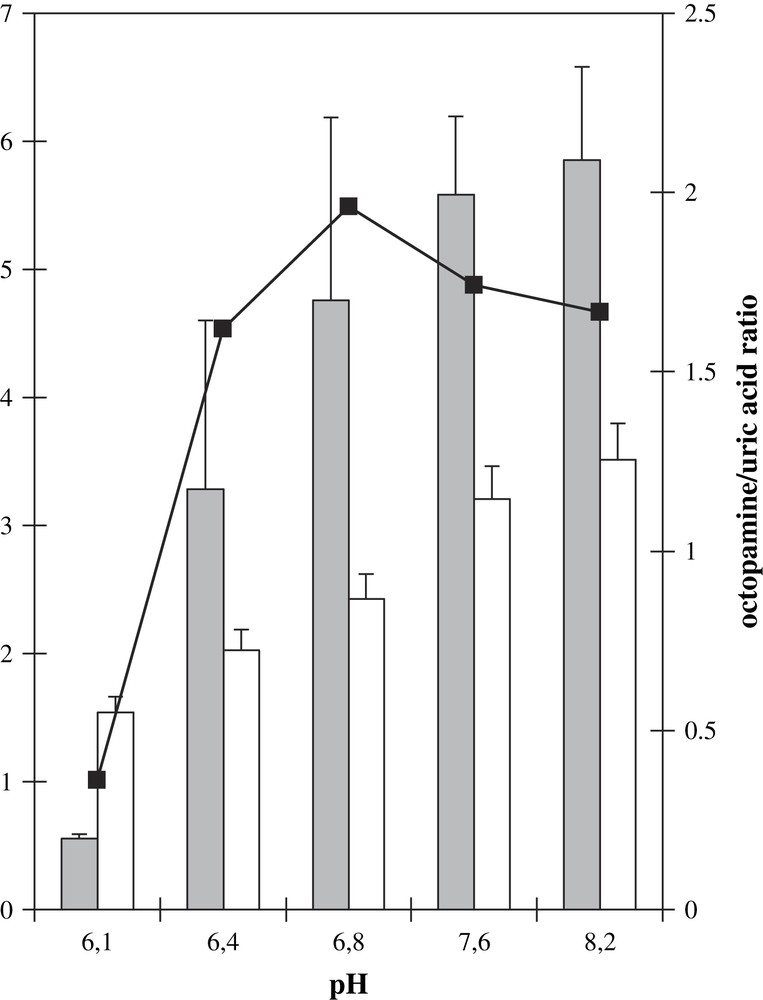
Octopamine (gray bars) and uric acid (white bars) formations as a function of pH. Samples contained DβM, 40 μg/mL, XOD, tyramine, xanthine, catalase and 50 mM phosphate buffer. For activities at pH 6.1 and 6.4, acetic acid was introduced to final concentrations of, respectively, 10 mM and 5 mM. Bars: raw octopamine formation measured after HPLC chromatograms (μM). Values are average of three experiments ± standard deviation. Squares: ratio of the amount of octopamine formed (μM) vs. that of uric acid (mM × 10) at the same pH.
3.4 Dopamine β-monooxygenase
The influence of the amount of DβM in the reaction mixture was also investigated. This study was performed over a range of 32–160 μg/mL in DβM concentrations, all other parameters being the same as in the previous experiments. A classical system would produce a linear dependence of product formation as regards to the amount of enzyme (Fig. 6). Activity was a linear function of added DβM when the enzyme concentration remained lower than 40 μg/mL. Increasing the amount of DβM in the buffer resulted in an octopamine production superior to that predicted by a linear extrapolation of the first points (activity of 4.6 nmol/min/mg for 40 μg/mL DβM concentration vs. 9.6 nmol/min/mg for 160 μg/mL). This result is consistent with the existence of a dimer–tetramer equilibrium. Saxena et al. observed a change in the dimer–tetramer equilibrium constant from 1.8 × 10−6 M to 1.16 × 10−9 M when the pH is increased from 5.0 to 5.7 [26]. Since in our conditions pH is 7.6, we assume that the main form is the dimer and an increase of enzyme concentration displaces the equilibrium towards the more active tetrameric form [27].
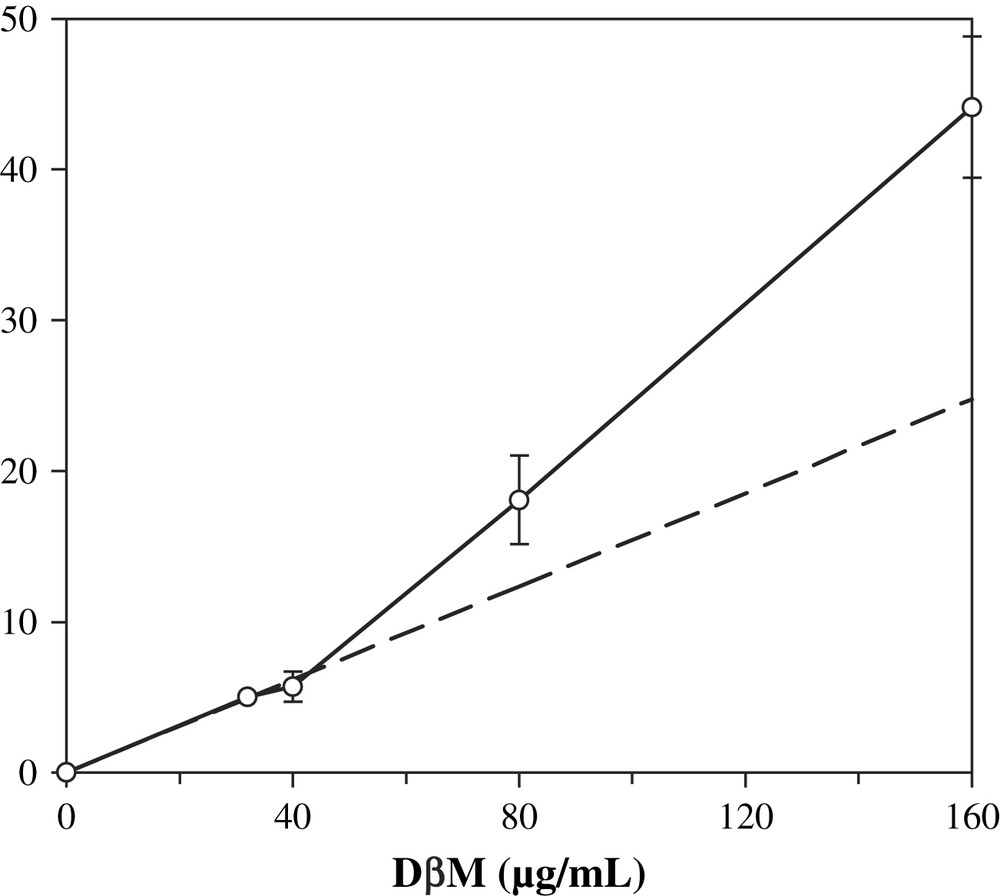
Influence of the concentration of DβM on octopamine formation. Conditions are similar to standard assay, except that DβM concentrations varied from 32 to 160 μg/mL. Dotted line is an extrapolation of values for 0, 32 and 40 μg/mL DβM concentrations. Values are average of three experiments ± standard deviation.
3.5 XOD dependence
XOD influence on activity was studied. By varying this enzyme amounts in the activity buffer, we could show a dependence on octopamine formation. Increasing XOD quantities from 0.2 mU to 2.5 mU, under a constant xanthine concentration, led to increasing quantities of product after incubation with DβM (Fig. 7). Uric acid production also varied, but its production remained a linear function of XOD quantities. As for octopamine formation, the increase was higher at low XOD unit values (0.2 to about 1 mU) and displayed a slower variation at higher values. Nevertheless, the specific activity obtained at 2.5 mU XOD is higher than the value obtained at 1 mU (6.0 vs. 4.6 nmol/min/mg). Therefore the activity of DβM does not parallel that of XOD when varying the amounts of the latter, and a better specific activity for DβM was obtained using higher amounts of XOD.
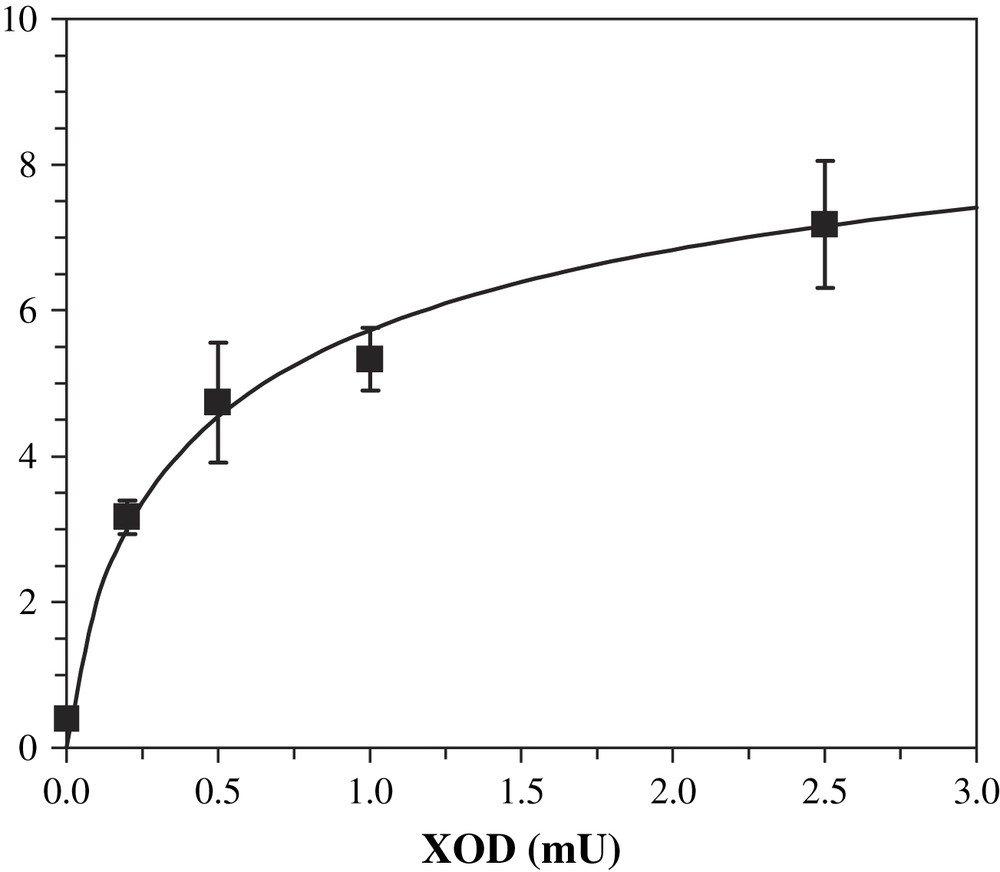
Influence of the amounts of XOD on the production of octopamine by DβM. Conditions are similar to standard assay, except for the amount of XOD. Each point is the average of three experiments ± standard deviation.
The amount of superoxide produced by the XOD system was deduced from the final concentration of uric acid. The ratio between superoxide and uric acid was obtained after assuming an exponential dependence with pH of that ratio, using the values determined by Fridovich [23] at pH 7.0 and 10.0. Lineweaver–Burke plot for the dependence of octopamine formation as versus produced superoxide yielded the following values, assuming a Michaelis and Menten mechanism (linear regression coefficient R2 = 0.98): and , i.e. kcat = 0.87 min−1. Thus the very low VM value for the XOD-assisted activity of DβM seems to be the parameter that would explain such a low activity, compared to that obtained with ascorbate as a cofactor. It should be considered that these values are not the real parameters for the kinetics that is studied here, because the system probably proceeds through a more complex pathway as a simple Michaelis and Menten kinetics.
3.6 Substrate concentration
An amount of 1 mU XOD was used in most experiments. Species concentrations were varied from this point, in order to further establish the influence of each ingredient onto activity. We have seen that superoxide is present in limited quantities at such concentrations, for lower quantities of XOD produce a lower activity (Fig. 7). The influence of the second cofactor, tyramine, was also investigated. At a constant pH of 7.6 and 1 mU XOD, substrate concentration was diminished down to 0.1 mM, and octopamine formation was unchanged (data not shown). Therefore we believe that tyramine recognition by DβM with superoxide anion as a cofactor is a favorable phenomenon, and that the steps involving superoxide are kinetically limiting, as suggested by the low value calculated for superoxide.
4 Discussion
The ability of a molecule to be a reducer for DβM is linked with its redox potential and that of DβM copper ions. Therefore we shall comment both on the redox potential of DβM and on that of some known reducers. No direct experiment could establish the oxidoreduction potential for the copper atoms of DβM. However, non-direct measurements have suggested an average value for both ions. Walker et al. [5] measured a value of +310 mV at pH 7.0 (E∗ = redox potential/NHE at pH 7.0) using EPR spectroscopy, while Ljones and Flatmark [28] determined an E∗ of +385 mV. Ferrocyanide, a DβM reducer [28], possesses a redox potential close to +410 mV. The ability of its oxidized form to oxidize DβM suggests that E∗ for DβM is close to that of that couple [5]. Along with EPR measurements, this result confirms the fact that the E∗ for DβM is about +400 mV. Since E∗ for O2/O2− is −160 mV, superoxide anion is consequently a good candidate for the reduction of DβM. Moreover, E∗ for O2−/H2O2 is +890 mV. A SOD activity is favored by an E∗ comprised between +200 and +500 mV [29]. As having an E∗ of about +400 mV, DβM is likely to possess an SOD activity as regards to thermodynamics.
Using the Cyt. c assay, a low SOD activity was measured for DβM (about 3.7% of that of bovine erythrocytes CuZn–SOD). The lower activity obtained for DβM can be understood because: (i) with the presence of a bridging imidazole which confers original redox properties [30] and (ii) a guiding of the superoxide anion down to the active site through a channel of charged residues [31]; CuZn–SOD is designed to react with superoxide. In DβM, the tertiary structure of the amino acids (in the surroundings of the copper ions) is unknown. We propose that the lower SOD activity of DβM may be due to the organization of the amino acids in the vicinity of the active site. If the copper ions are located on the surface of the protein and exposed to solvent, as observed for PHM [10], the copper ions of DβM would possess a lower reactivity towards an unspecific anion like superoxide. Superoxide might thus be more available for disproportionation or attack by other molecules in DβM than in CuZn–SOD.
By varying the pH of the activity buffer from 6.1 to 8.2, we observed an important increase of DβM activity (Fig. 5). Since we also observed an increase in uric acid production, we could have assumed that the pH dependence of DβM activity is only due to an increase of the concentration of superoxide anion. However, the pH dependence in uric acid production and consequently superoxide is not as important as that of octopamine. From pH 6.1 to 8.2, we observed an increase of 2.3 in uric acid production vs. 10.6 in octopamine production. The ratio of octopamine to uric acid produced showed a maximal value at a pH of 6.8.
These results can be paralleled with those obtained when ascorbate is used as a cofactor. Miras-Portugal et al. [32] studied the activity of DβM as a function of pH in the range of 4.5–7.5. They observed an increase in pH from 4.5 to 4.8 followed by a decrease in pH to 7.5. These two variations were attributed to deprotonation of important active site residues. Concerning the decrease at pH higher than 4.8, the authors proposed that a histidine residue may be involved in catalysis, whose side-chain proton abstraction would prevent activity. As for the increase in activity at lower pH values, the authors proposed the involvement of a carboxylate side-chain, whose deprotonation would favor an electrostatic interaction with protonated amine substrate. Nevertheless, neither of these residues was characterized by later studies.
CuZn–SOD was also shown to possess a pH-dependent activity. Getzoff et al. measured a sharp decrease in activity at pH superior to 9.5 [31]. Using site-directed mutagenesis, three residues were later characterized as being responsible for such a dependence in Xenopus laevis, Lys120, Lys 134 and Arg141 [33]. Arg141 is located near the active site copper, and Lys120 and 134 are involved in a hydrogen bond network between the surface of the protein and copper. These three positively charged residues are involved in the guidance of superoxide anion towards the copper site.
We can interpret our pH study results in the light of both previous studies. Difference in activity from pH 6.1 to 6.8 can be attributed to the involvement of a basic active site residue, whose deprotonated state is the only catalytic one. For instance, this residue could be one that favors docking of catecholamine or amine substrate by electrostatic interaction. The very low decrease at higher pH values, as opposed to the sharp decrease described when ascorbate is used as a cofactor [32], lets us assume that no positively charged residue with a pKa lower than 8 is involved in catalysis. That is to say, only a Lys or Arg residue could be responsible for an eventual decrease in activity at higher pH values, whereas involvement of a histidine residue is quite unlikely.
The absence of a positively charged residue nearby the active site would further confirm our earlier assumptions. Indeed, when comparing the three-dimensional structure of CuZn–SOD with that of DβM, we suggested that in DβM, no positive residue is located near CuM, thus disfavoring electrostatic guidance of superoxide. Analysis of pH-dependent activity is in agreement with this assumption. Therefore our study enables us to make new proposals as regards to the geometry of the active site of DβM, and the results obtained from the various experiments performed here are in good agreement.
5 Conclusion and mechanistic proposal
We demonstrated that DβM is able to catalyze the hydroxylation of substrate tyramine in the presence of superoxide anion generated by the xanthine/xanthine oxidase system. We shall now comment on the formation of the copper–oxygen intermediate responsible for the hydroxylation of tyramine.
We observed that in the absence of ascorbate, DβM exhibits a SOD-like activity. We therefore propose the reduction of copper by superoxide as a first step. Either copper ion could be reduced in that step to give mixed valence species CuHI/CuMII or CuHII/CuMI, which, after a second reduction, can lead to the fully reduced form CuHI/CuMI. These pathways suggest differences in the reactivity of each copper ion of DβM towards superoxide ion. The present study does not establish any distinction in the reactivities of both copper ions of the enzyme. In other words, we could not determine whether only one of the copper ions at the active site of DβM or the two of them are reduced by superoxide and, in the second case, whether it was substrate binding CuM or CuH that reacted more easily with this anion. Nevertheless, by considering the exposure of the copper ions of PHM to solvent, we can assume that no channel preferentially drives O2− towards either copper ion of the active site. Therefore we may assume that both site exhibits similar reactivity regarding superoxide, and that the fully reduced form CuHI/CuMI, which is the form able to bind the substrate, is rapidly formed [34]. Now two ways can be considered. First of all, the classical reaction of the fully reduced DβM with substrate and dioxygen which leads to the catalytic reaction. In this mechanism, the role of superoxide anion is only restricted to the role of reducing agent (Scheme 4a). However, the very low activity for the XOD-assisted activity (kcat = 0.87 min−1) compared to that obtained with ascorbate (kcat = 120 min−1) as a cofactor suggests another role to superoxide and that the system probably proceeds through a more complex pathway.
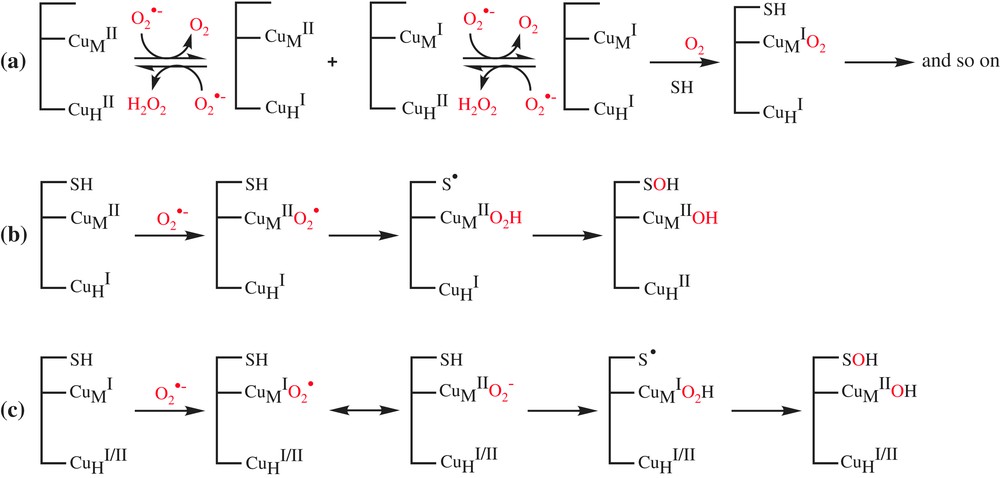
Proposed mechanisms for the superoxide-assisted DβM activity at the copper centers.
The second pathway involves the addition of a superoxide molecule at CuMII of the preformed complex of mixed valence species CuHI/CuMII with the substrate, followed by a H-atom abstraction to substrate according to the mechanism proposed by Klinman et al. [17] (Scheme 4b). This mechanism, which involves the complexation of substrate to semi-oxidized form of DβM, is unlikely, since the substrate is known to have a good affinity for the fully reduced form of enzyme [1]. Another pathway is the addition of a superoxide molecule at CuMI of the preformed complex of the fully reduced CuHI/CuMI or the mixed valence CuHII/CuMI forms with the substrate (Scheme 4c). The resulting reduced oxygen species CuMIO2↔CuMIIO2− abstracts substrate H-atom according to the mechanism proposed by Prigge et al. [18].
While the pathway c seems more likely because it involves a pre-catalytic complex of substrate with a fully or semi reduced enzyme, it is difficult to decide in favor of one of these pathways. We believe that a way to clearly address the selectivity of superoxide vs. the copper ions of DβM, and to establish the pathway for the superoxide-assisted activity of DβM would be to prepare a semi-depleted enzyme.