

1 Introduction
Galactose oxidase (GOase) is a copper-dependent metalloenzyme which converts primary hydroxyl functions into aldehydes with high turn-over frequencies of about 800 s−1 (2.8 × 106 h−1) [1]. The enzyme is very selective and secondary hydroxyl groups are not converted. A wide range of different primary alcohols can be employed as substrates. Extensive investigations with the enzyme and model complexes [2–8] allowed obtaining a rather precise picture of the mechanism. In simplified form this consists in: (i) substrate (=alkoxide) binding to a Cu(II) tyrosinyl radical complex to give a [Cu(II)(OTyr)(OCH2R)] complex, (ii) intra-molecular H-transfer (the rate determining step) to generate a ketyl anion complex [Cu(II)(HOTyr)(O-CHR)], (iii) intra-molecular oxidation of the ketyl radical anion to give [Cu(I)(HOTyr)] and the product OCHR, and (iv) oxidation of [Cu(I)(HOTyr)] with molecular oxygen, O2, to regenerate the initial tyrosinyl radical complex [Cu(II)(OTyr)] and H2O2. With EtOH as a substrate, the reaction EtOH + O2 → MeCHO + H2O2 is exothermic by about −25 kcal mol−1.
Many transition-metal catalysts have been developed over the last decades, which are capable of converting alcohols into the corresponding carbonyl compounds [9], and the combination of catalytic amounts of (Pr4N)+[RuO4]− (TPAP) with N-methylmorpholine as oxidant proved to be very useful [10]. Especially noteworthy are catalysts derived from Pd(OAc)2/pyridine (Uemura catalyst) or Pd(OAc)/phenanthroline (Sheldon catalyst), which use O2, air, or H2O2 as terminal oxidants and can be even used in aqueous solutions [10–12]. Mostly the classical mechanisms frequently encountered in organometallic chemistry were proposed [12a]. A recent contribution discusses the α-hydrogen atom transfer to a coordinated tetramethyl piperdineoxide (TEMPO) radical from a copper-bonded alkoxide as the key step in alcohol oxidation [13].
With the objective to find organometallic radical complexes which are like Goase able to selectively convert primary hydroxyl functions into the corresponding aldehydes, we started to investigate rhodium and iridium aminyl radical complexes. We chose rhodium and iridium as metals because the bond energies of ligand atoms to these metals are intrinsically high and consequently the catalysts should be rather stable [14]. Aminyl radicals, NR2, were selected because they offer the possibility to better control the steric environment of the complex and especially to construct polydentate chelate ligands which additionally enhance the stability of the complexes. Indeed, the persistent Rh(I) aminyl radical cation [Rh(trop2N)(bipy)]OTf I [15] and a neutral aminyl radical rhodium complex to which was assigned the structure [Rh(trop2dach-2H)] II [16] were isolated (Scheme 1).
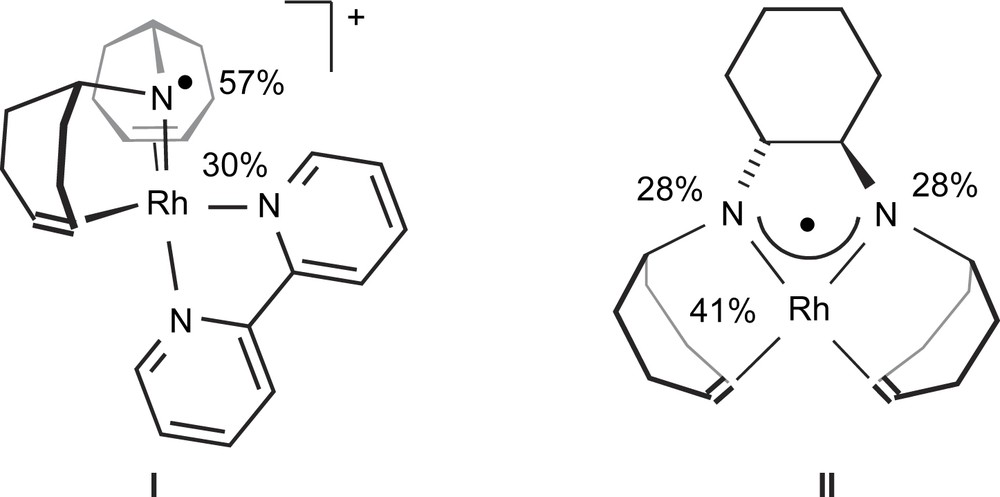
Structures of rhodium aminyl radical complexes I and II.
These complexes contain chelating aminyl radicals as ligands which were constructed on the basis of the dibenzo[a,d]cyclohepten-5-yl platform (in the following we will refer to the trivial name dibenzotropylidenyl and use the abbreviation “trop” for this group). Pulse EPR spectroscopy in combination with DFT calculations allowed us to assign the spin distribution in both complexes. In I, 30% of the spin density is located at the Rh-center and 57% at the N one. In II we find 41% at Rh and 2 × 28% at both nitrogen centers. In accordance with these findings, both I and II abstract hydrogen from activated hydrogen donors (stannanes, R3SnH, thiols, RSH, and in case of II silanes, R3SiH) and the corresponding amine complex [Rh(trop2NH)(bipy)]OTf and amino amide [Rh(trop2dach-1H)] are formed almost quantitatively. But the abstraction of α-hydrogens from alcohols was not observed. Interestingly, with the corresponding neutral iridium aminyl radical complex [Ir(trop2dach-2H)] (4) and 1,4-benzoquinone (BQ) as hydrogen acceptor the catalytic dehydrogenation of primary alcohols to the corresponding aldehydes was observed [17].1 This reaction proceeds with unprecedented high activity (TOFs > 150,000 h−1) even for non-activated alcohols and selectivity, that is secondary hydroxyl groups are not converted.
In this report we discuss the properties and structures of the iridium complexes [Ir(trop2dach)]OTf (1) and [Ir(trop2dach-1H)] (2), which are likely to be involved in the catalysis. A comparison is made with the previously reported anionic bis(amido)complex [K(18-C-6)][Ir(trop2dach-2H)] (3) [17]. The highly efficient catalytic dehydrogenation of naturally occurring primary alcohols was investigated in order to evaluate the scope of the iridium aminyl radical-catalyzed dehydrogenation.
2 Results
2.1 Properties of iridium trop2dach complexes 1, 2, and 3
The iridium complexes 1 and 2 were prepared as outlined in Scheme 2. The one-step synthesis of 3 from 1 has been reported before [17].

Syntheses of complexes 1–3.
The neutral amino amide complex [Ir(trop2dach-1H)] (2) is obtained from 1 by deprotonation with sodium methanolate in methanol. Under these conditions, double deprotonation to the di(amido) complex 3 is not observed. The di(amido)iridate [K(18-C-6)][Ir(trop2dach-2H)] (3) is obtained when 2 is treated with the stronger base KOtBu/18-crown-6 in THF. In course of the deprotonation the absorption of the longest wave length, λmax, is successively red-shifted from 486 nm in 1 to 504 nm in 2 to 524 nm in 3. We determined the pKa values for each deprotonation step using a classical 1H NMR spectroscopic technique frequently used for organic molecules [18]. For each equilibrium (1) and (2),
| [Ir(trop2dach)]+ + Ref(1)− ⇌ [Ir(trop2dach-1H)] + RefH(1) | (1) |
| [Ir(trop2dach-1H)] + Ref−(2) ⇌ [Ir(trop2dach-2H)]− + RefH(2) | (2) |
Like the corresponding rhodium complex [Rh(trop2dach)]OTf [19],2 the iridium complex 1 is reduced in two consecutive one-electron steps, at E½1 = −1.87 V to the metallo radical [Ir(trop2dach)] and at E½2 = −2.52 V to the iridate complex [Ir(trop2dach)]− [all potentials vs. ferrocenium/ferrocene (Fc+/Fc) as reference, DMSO as solvent, Pt working electrode, scan rate = 100 mV s−1). No oxidation below +1 V was observed. Although we could not measure the potentials for the oxidation reactions, [Ir(trop2dach-1H)] (2) − e → [Ir(trop2dach-1H)]+ and [Ir(trop2dach-2H)]− (3) − e → [Ir(trop2dach-2H)], with sufficient certitude, very likely they occur at low oxidation potentials. The analogous rhodium complexes are reversibly oxidized at remarkably low potentials: [Rh(trop2dach-1H)] at E½ = −0.34 V [19], and the di(amido) rhodate [Rh(trop2dach-2H)]− at E½1 = −1.02 V to give the radical [Rh(trop2dach-2H)], which is again reversibly oxidized in a second step still at a low potential, E½ = −0.38 V [16,20].
Crystals suitable for X-ray diffraction of (R,R)-[Ir(trop2dach)]OTf 1 were obtained from a concentrated THF solution layered with n-hexane and contain two THF molecules in the crystal lattice. Crystals of (S,S)-[Ir(trop2dach-1H)] (2) with the chiral space group P212121 were grown from a concentrated diethylether solution layered with n-pentane. They stem from one experiment where we used the racemic mixture of the trop2dach ligand; that is the crystals exist in two enantiomorphic forms. In all other experiments, the enantiomerically pure (R,R)-trop2dach was employed as ligand. Plots of the structures of 1 and 2 are shown in Figs. 1 and 2, selected bond lengths and angles are given in the figure captions and pertinent crystal data are given in Table 1 in the experimental section. In 1 and 2, the iridium center is embedded in a tetrahedrally distorted planar coordination sphere as is indicated by the angle φ given by the intersection of the N1–Ir–ct1 and N2–Ir–ct2 plane, which amounts to 20°. There is remarkable little change of the bond lengths and angles when 1 is deprotonated to 2. One would expect that the amide nitrogen center, N1 in 2, exerts a trans-influence leading to a lengthening of the distances to the opposite ligands [21,22]. This is not the case. The distances between the iridium atom and the centroids, ct, of the coordinated CCtrop groups remain almost constant, at about 2.0 Å. Obviously, the CCtrop units serve as a kind of “electronic buffer” and absorb the electron density donated by the amide groups by efficient metal-to-ligand back-donation [that is the increased electron population in the filled d-orbitals at the iridium center is depleted by donation into the π∗ orbitals of the coordinated CCtrop groups, d(Ir) → π∗(CCtrop)].
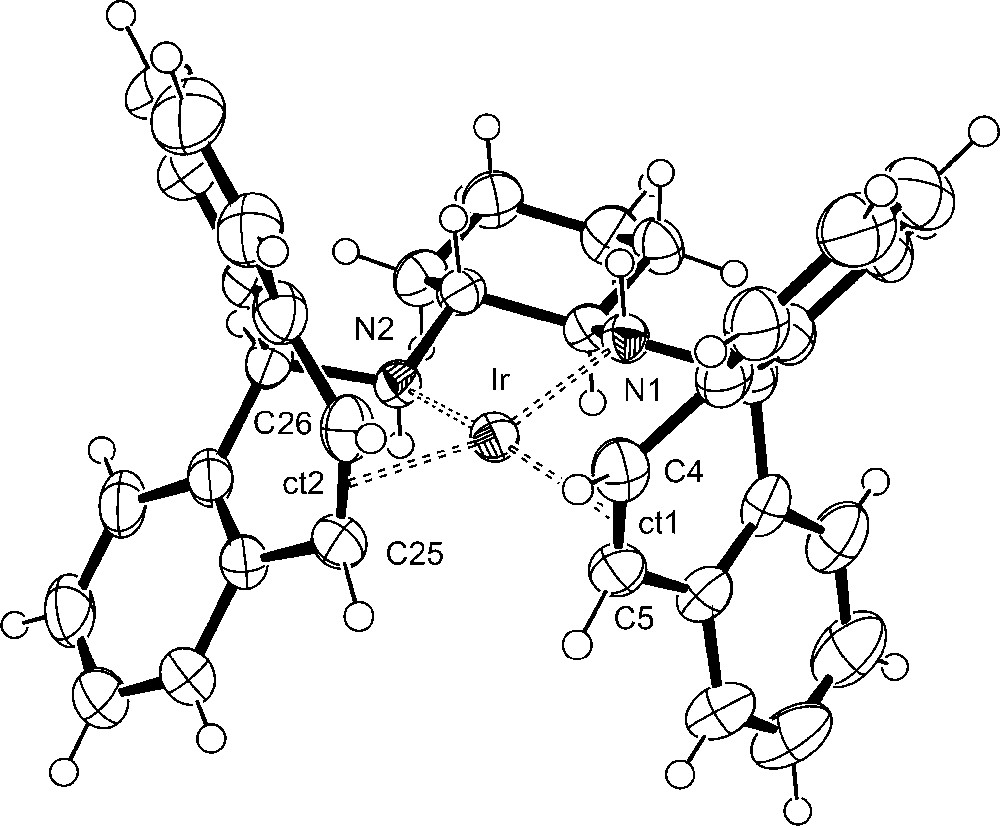
Plot of the structure of the cation in (R,R)-[Ir(trop2dach)]Otf (1). The disordered triflate anion and THF molecules are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ir–N1 2.095(3), Ir–N2 2.090(3), Ir–ct1 1.986(4), Ir–ct2 2.004(4), C4–C5 1.421(6), C25–C26 1.401(6); ∑(N1) 339.6, ∑(N2) 340.9. The angle φ, defined by intersection of the N1–Ir–ct1 and the N2–Ir–ct2 plane, amounts to 20.6°.

Plot of the structure of (S,S)-[Ir(trop2dach-1H)] (2). The pentane molecule, incorporated in the unit cell, is omitted for clarity. Selected bond lengths [Å] and angles [°]: Ir–N1 1.995(5), Ir–N2 2.143(5), Ir–ct1 2.026(6), Ir–ct2 2.026(6), C4–C5 1.448(9), C25–C26 1.468(9); ∑(N1) 353.0, ∑(N2) 338.7. The angle φ, defined by intersection of the N1–Ir–ct1 and the N2–Ir–ct2 plane, amounts to 20.1°.
Crystal data and refinement of compounds 1, 2, and 3
| Compound | 1 | 2 | 3 |
| Empirical formula | C45H50F3IrN2O2S | C41H45IrN2 | C56H72IrKN2O8 |
| M | 980.13 | 757.99 | 1132.46 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P21 | P212121 | P21 |
| a/Å | 9.3769(6) | 13.075(2) | 11.7856(7) |
| b/Å | 17.870(1) | 15.655(3) | 24.750(1) |
| c/Å | 12.6318(8) | 17.739(3) | 17.254(1) |
| α/° | |||
| β/° | 103.627(1) | 95.342(1) | |
| γ/° | |||
| V/Å3 | 2057.1(2) | 3631(1) | 5010.9(5) |
| μ/mm−1 | 3.358 | 3.706 | 2.805 |
| Dcalcd/g cm−3 | 1.582 | 1.387 | 1.501 |
| Crystal dimensions/mm | 0.23 × 0.20 × 0.18 | 0.17 × 0.15 × 0.14 | 0.31 × 0.23 × 0.21 |
| Z | 2 | 4 | 4 |
| T/K | 298 | 200 | 200 |
| 2θmax/° | 56.64 | 55.26 | 56.58 |
| Refls. measured | 21,460 | 20,454 | 69,595 |
| Refls. unique | 10197 (Rint = 0.0216) | 7665 (Rint = 0.0608) | 24616 (Rint = 0.0304) |
| Parameters/restraints | 587/33 | 397/38 | 1225/14 |
| R1 [I ≥ 2σ(I)] | 0.0267 | 0.0376 | 0.0302 |
| wR2 (all data) | 0.0636 | 0.0895 | 0.0685 |
| Max./min. res. elec. dens./e Å−3 | 1.089/−0.399 | 1.499/−1.375 | 1.611/−0.502 |
In line with this interpretation, the coordinated CC bond trans to the amide nitrogen N1 is slightly lengthened in 2. The Ir–N bonds become slightly shorter upon deprotonation and shrink below a length of 2.0 Å while the Ir–amine bonds, Ir–NH, are slightly longer than 2.0 Å. Note that the sum of the C–N–C and two Ir–N–C bond angles, ∑(N), becomes significantly larger by about 13° when the NH function is deprotonated, but the amide nitrogen still has a slightly pyramidal coordination sphere [∑(N1) = 353.0°]. The discussed structural trends hold also for the diamido complex [Ir(trop2dach-2H)]−, that is the Ir–N bonds are <2.0 Å, the coordinated CCtrop units elongated, and ∑(N1) ≈ 353°. Only the complex adopts a lesser distorted structure closer to a planar one (φ = 8.1°).
In Table 2, the 1H NMR and 13C NMR data of the olefinic protons and carbon nuclei of the coordinated CCtrop groups of 1 and 2 are given. The last column shows the chemical shifts of the NH protons of the coordinated amine functions. For comparison we list also the corresponding data for the diamidoiridate 3 and the rhodium complexes [Rh(trop2dach)]+, [Rh(trop2dach-1H)], and the anion [Rh(trop2dach-2H)]−.
1H and 13C NMR resonances [ppm] of the CHCHtrop units and the NH function in 1, 2, and 3
| δ 1Hol | δ 1Hol | δ 13Col | δ 13Col | δ 1H(N) | |
| [Ir(trop2dach)]+ (1) | 4.02 | 4.75 | 60.4 | 67.5 | 5.92 |
| [Ir(trop2dach-1H)] (2) | 2.66a | 3.34a | 47.8a | 51.4a | – |
| 3.46 | 4.09 | 54.8 | 62.9 | 5.74 | |
| [Ir(trop2dach-2H)]− (3) | 2.63 | 2.91 | 49.8 | 52.5 | – |
| [Rh(trop2dach)]+ | 4.16 | 5.38 | 70.1 | 83.4 | 4.45 |
| [Rh(trop2dach-1H)] | 2.99a | 3.49a | 67.0a | 67.3a | – |
| 4.29 | 4.83 | 71.2 | 81.4 | 5.07 | |
| [Rh(trop2dach-2H)]− | 2.90 | 3.79 | 66.4 | 73.0 | – |
a Data for CCtrop unit trans to the amide nitrogen N1.
The data are fully in accord with the structures determined in the crystalline state. Due to the tetrahedral distortion and the chirality of the ligand backbone leading to C2-symmetric structures for 1 and 3, the 1Hol and 13Col resonances become pairwise inequivalent and two resonances are observed. The neutral amine amide complexes [M(trop2dach-1H)] are asymmetric and consequently four distinct signals are obtained. Upon deprotonation, both the 1Hol and 13Col resonances of the CCtrop bond opposite to the amide group are considerably shifted toward lower frequencies (that is more shielded than the CCtrop units, opposite to the NH amine functions). This finding bolsters the above-made statement that the CCtrop bonds function as electronic buffers and accept the increasing electron density at the metal centers in the deprotonated species. Note that generally the 1Hol and 13Col resonances are recorded at significantly lower frequencies in the iridium complexes when compared to the rhodium analogues. This strong shielding indicates that metal-to-ligand back-donation d(M) → π∗(CCtrop) is more effective in the iridium complexes. Consequently, the filled d-orbitals at the metal are polarized towards the CCtrop units and away from the occupied p-type orbitals at the amide nitrogen centers representing the amide lone-pairs. The destabilizing 2-center-4-electron d(M)–p(N) interaction is diminished. Hence, the amido complexes 2 and 3 become less destabilized when compared to their rhodium counterparts and consequently the acids conjugated with 2 and 3, namely the amine complexes 1 and 2, become more acidic. Indeed, [Ir(trop2dach)]+ (1) is five order of magnitude more acidic than [Rh(trop2dach)]+ and the NH function in [Ir(trop2dach-1H)] (2) is about 3–4 orders of magnitude more acidic than the one in [Rh(trop2dach-1H)]. This is also clearly reflected by the more deshielded 1H NMR shifts of the amine protons in the iridium complexes 1 and 2 (Table 2). We had previously used this argumentation based on avoided filled–filled repulsions which was introduced by Caulton [23] to qualitatively interpret the physico-chemical properties of a series of [Rh(trop2dach)] complexes.
2.2 Oxidation of natural primary alcohols
Based on the mechanism for GOase, we proposed a catalytic cycle [17] which is presented in Scheme 3 in simplified form. The cationic bis(amino) complex 1 is doubly deprotonated by a base (KOtBu) to give the anionic bis(amido) complex 3, which is subsequently oxidized by 1,4-benzoquinone (BQ) to the iridium aminyl radical complex 4 and the semiquinone radical anion (SQ−). Both 4 and SQ− were detected by EPR spectroscopy [17]. As with GOAse, it is possible that the alcohol RCH2OH (or more likely the corresponding alkoxide) is coordinated to the iridium center whereby the α-hydrogen is placed in proximity to the nitrogen centered radical. Intra-molecular H-atom transfer may occur and an Ir-coordinated ketyl radical anion is formed. Likely this is oxidized in an inter-molecular reaction by SQ− to give the product, RCHO, and catechol (Cat). It was found that a wide range of simple aliphatic alcohols, arenes and heterocyclic compounds with primary hydroxyl functions can be converted to the corresponding aldehydes.
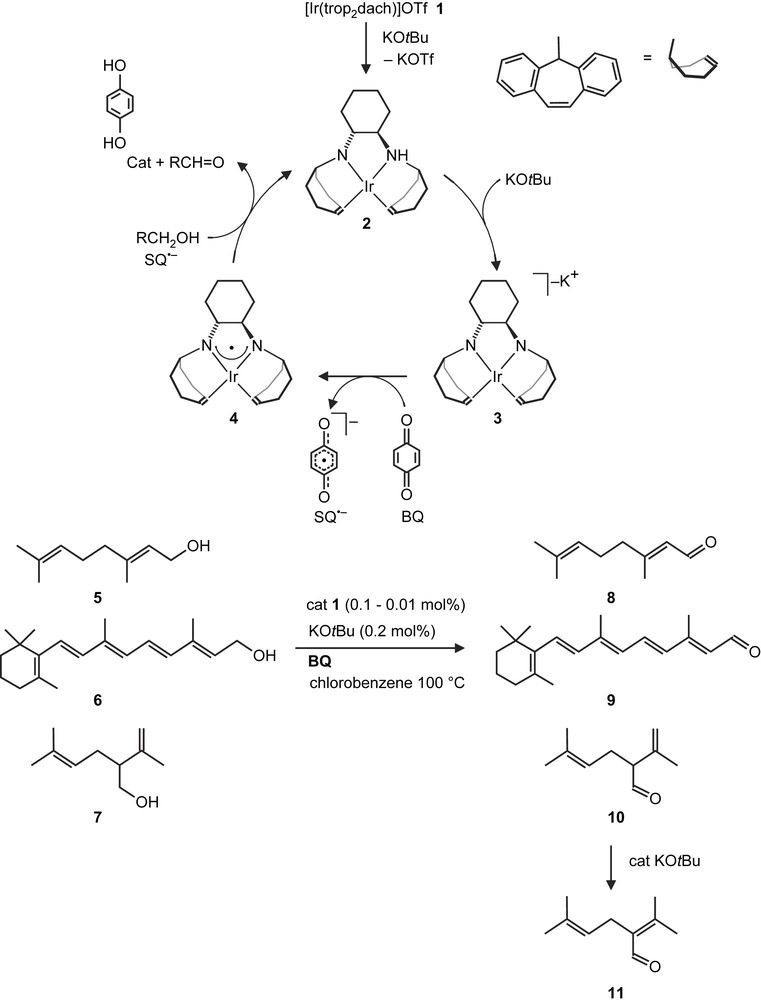
Proposed catalytic cycle for the conversion of the natural alcohols geraniol (5), retinol (6) and (±)-lavandulol (7).
We tested now some more complex naturally occurring alcohols as substrates in the catalytic dehydrogenation reaction. The reactions with geraniol (5), retinol (6) and (±)-lavandulol (7) were performed at 100 °C in chlorobenzene, which had proven to be a suitable solvent [17]. At a substrate-to-catalyst ratio S/C = 10,000 (0.01 mol% catalyst loading), complete and clean conversion of 5 and 6 to geranial 8 and retinal 9, respectively, were observed within minutes (Tables 3 and 4). The sterically more congested alcohol (±)-lavandulol 7 was much slower converted. At S/C = 10,000, not more than 30% of the alcohol was consumed, even after longer reaction times.
Catalytic dehydrogenation of geraniol (5), retinol (6) and (±)-lavandulol (7)
| Substrate | Product(s) | Yield | Time |
| Geraniol 5 | Geranial 8 | >98% | 20 min |
| Retinol 6 | Retinal 9 | >98% | 5 min |
| (±)-Lavandulol 7 | (±)-Lavandulal 10 | cf Table 4 | |
| Isolavandulal 11 |
Dehydrogenation of (±)-lavandulol 7 under various conditions
| S/C ratio | KOtBua (equiv) | BQb (equiv) | Time (h) | Yield (%) | Ratio 10:11 | |
| 1 | 1000 | 22 | 1.33 | 21 | 76 | 68:32 |
| 2 | 1000 | 66 | 1.33 | 19 | 57 | 0:100 |
| 3 | 1000 | 22 | 2.66 | 4 | 90 | 94:6 |
| 4 | 5000 | 44 | 2.66 | 168 | 57 | 7:93 |
a Equivalents of base with respect to catalyst.
b Equivalents of BQ with respect to substrate.
Therefore the S/C ratio was lowered to 1000 and under these conditions, comparable higher conversions were observed. However, we observed the formation of the isomerized product isolavandulal 11 beside (±)-lavandulal 10, which is clearly indicated by the singlet resonance of the aldehyde proton, CHO, at δ = 10.1 ppm beside the doublet at δ = 9.5 ppm, typical of the CHO of (±)-lavandulal 10. Isolavandulal 11 was isolated from the reaction mixture, purified and identified by comparison with the 1H NMR data reported in the literature [24,25].
To study the parameters that influence the formation of (±)-lavandulal 10 and isolavandulal 11, the catalytic oxidation of (±)-lavandulol was performed under varying conditions: 0.1 mol% (entries 1–3) and 0.02 mol% (entry 4) of the catalyst precursor 1 were used, the catalytic amount of KOtBu was varied between 0.22 and 0.66 mol%, and the amount of BQ was varied from 1.33 to 2.66 equivalents with respect to lavandulol 7.
Clearly, the amount of isolavandulal 11 increases with increasing base concentration (compare entries 1 and 2) and with increasing reaction times (entry 4). With smaller amounts of base and larger concentrations of BQ, about 90% of total conversion to aldehydes 10 and 11 is obtained and (±)-lavandulal 10 can be obtained as the main product (entry 3). On the other hand, with low catalyst loadings, longer reactions times, and relatively high base concentration, the main product is isolavandulal 11 (entry 4).
The evolution of the ratio of 10:11 was followed with time under otherwise identical conditions (0.1 mol% catalyst, 66 equiv KOtBu, 1.33 equiv BQ). The result is displayed in Fig. 3, which clearly shows that the total conversion to aldehydes 10 and 11 does not significantly change while increasing amounts of isolavandulal 11 are formed. The experiments thus strongly indicate that the isomerization 10 to 11 is a classical base-catalyzed reaction and is not influenced by the metal complex.
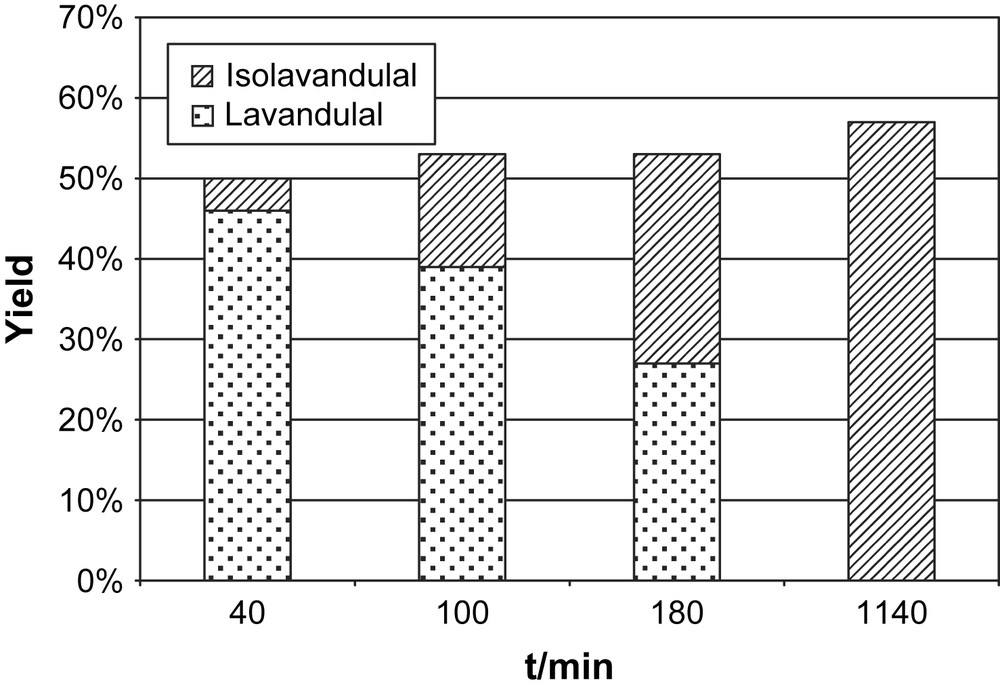
Time-dependent isomerization of (±)-lavandulal 10 to isolavandulal 11 after dehydrogenation of (±)-lavandulol 7. Reaction conditions: catalyst loading 0.1 mol%, 0.66 mol% KOtBu, 1.33 equiv BQ with respect to 7.
3 Conclusion
In conclusion, it was shown that the iridium trop2dach complexes have very similar ground-state structures compared to their rhodium counterparts. Remarkably, the iridium complexes are significantly more acidic, which can be explained by a stronger metal-to-olefin back-donation, which especially stabilizes the amido complexes. These are relatively easily oxidized and a metallo radical like [Ir(trop2dach-2H)] 4 is very likely involved in the very efficient catalytic conversion of natural primary alcohols like geraniol and retinol to the corresponding aldehydes, geranial and retinal. Sterically more congested alcohols are converted at lower rates. This suggests that the exclusive conversion of primary alcohols to aldehydes has primarily steric reasons. We believe that our observation that metal-coordinated radicals can be used for highly efficient and selective reactions is an important metalloenzyme-inspired finding, which may lead to the development of new types of catalysts.
4 Experimental part
4.1 General procedure
All manipulations of air or moisture sensitive compounds were performed on a standard vacuum line in flame-dried glassware under an atmosphere of argon (purified with an MBraun 100 HP gas purification system). Solvents were distilled under argon from sodium/benzophenone (THF, hexane), calcium hydride (acetonitrile), and phosphorus pentoxide (chlorobenzene). Air-sensitive compounds were stored and weighed in a glove-box (M Braun: Labmaster 130 or 150B-G). Reactions performed with small quantities of compound were performed in a glove-box.
NMR measurements were carried out on Bruker Avance 200, 250 and 300 MHz spectrometers at room temperature. Chemical shifts δ are given by definition as dimensionless numbers. The absolute values of the coupling constants are given in hertz (Hz), regardless of their signs. The assignments were accomplished using multi-dimensional methods. Multiplicities are abbreviated as singlet (s), doublet (d), triplet (t), quartet (q), and broad (br). Spectra were referenced with external standards: for 1H and 13C NMR with TMS and for 19F NMR with CFCl3.
UV/vis spectra were recorded on a UV/vis/NIR lambda 19 spectrometer in 5-mm quartz cuvettes (200–1000 nm).
Gas chromatography was performed on a Hewlett Packard HP 6890 Series GC system equipped with a HP-5 crosslinked 5% PH ME siloxane column (30 m × 0.32 mm, film thickness 0.25 μm), flow rate 27.2 mL/min at 1.42 psi. The following temperature program was used: initial temperature 80 °C (hold 1 min), increase to 180 °C at a rate of 4 °C/min and hold for 40 min.
Cyclic voltammetric investigations were performed using a Princeton Applied Research potentiostat/galvanostat model 263A. Working electrode: planar platinum electrode (approximate surface area 0.785 mm2); reference electrode: silver; counter electrode: platinum wire. A 0.1 M solution of nBu4NPF6 in dimethylsulfoxide (DMSO) was used as electrolyte. A scan rate of 100 mV s−1 was used and at the completion of each measurement, ferrocene (Fc) was added as an internal standard for calibration.
(R,R)-[Ir(trop2dach)]OTf (1): 300 mg [Ir2(μ-Cl)2(coe)4] (0.34 mmol) are dissolved in 25 mL THF and 331 mg (0.68 mmol) trop2dach and a few drops of CH3CN are added. To this red solution 172 mg (0.68 mmol) of silver triflate is added and a white solid precipitates. After stirring for 1 h at r.t., the reaction mixture is filtered through Celite. All volatiles are removed in vacuo and the raw product is crystallized from THF/hexane (1:1), yielding 259 mg (309 mmol, 91%) of the product as red crystals. After decanting the solvent, the product is thoroughly dried in vacuo. For 1H NMR and 13C NMR data of diagnostic value, see Table 2. – 19F NMR (188.3 MHz, d8-THF): δ = −80.7 (s, OTf). UV/vis (THF): λ [nm]: 486, 397, 306.
(R,R)-[Ir(trop2dach)-1H] (2): To a solution of 180 mg of 1 (0.22 mmol) in methanol, sodium methanolate is added. The color changes to dark red and all volatiles are removed in vacuo. The raw product is dissolved in diethyl ether and stirred for 20 min. After filtering, the precipitate of the solution is layered with hexane and red crystals grow overnight to yield 129 mg (0.19 mmol, 86%) of the product. For 1H NMR and 13C NMR data of diagnostic value, see Table 2. – UV/vis (THF): λ [nm]: 504, 423, 280.
(R,R)-[K(18-C-6)][Ir(trop2dach)-2H] (3): To a solution of 100 mg of 1 (0.12 mmol) in THF, 29 mg KOtBu (0.26 mmol) and 32 mg 18-C-6 (0.12 mmol) are added. The color changes to dark red and the solution is layered with hexane. Red crystals grow overnight to yield a dark red powder, 101 mg (0.11 mmol, 94%). For 1H NMR and 13C NMR data of diagnostic value, see Table 2. – UV/vis (THF): λ [nm]: 524, 355, 274.
Catalyses: The catalyst precursor was dissolved under stirring in chlorobenzene; reactions with a S/C ratio of 10,000 and 5000 were conducted in 25 mL solvent, 1000 in 6 mL solvent. First, KOtBu and then after 5 min alcohol was added. The mixture was heated to 100 °C and BQ was added in one portion. The reactions were then monitored by GC and 1H NMR.
4.2 Work-up procedure for lavandulyl aldehydes
The reaction mixture was filtered through Celite and the solvent removed in vacuo (45 °C, 40 mbar). 1H NMR spectra showed, according to the progression of the reaction, a mixture of the alcohol and the respective aldehyde(s). To further purify isolavandulal 11, the dark, oily residues were filtered through aluminum oxide and washed with chlorobenzene. Again, the solvent was removed in vacuo. The aldehyde 11 was then obtained as an oil, contaminated with traces of quinones and therefore still rather dark in color. 1H NMR data of 11 were found to be in agreement with the literature [19,20].
4.3 X-ray diffraction studies: details of the data collection and refinement for 1, 2, and 3
Crystals suitable for X-ray analysis were mounted in degassed perfluoropolyalkylether on top of a glass fiber and then brought into the cold nitrogen stream of a low temperature device so that the oil solidified. Data collection for the X-ray structure determinations were performed on Bruker SMART Apex diffractometer system with CCD area detector by using graphite-monochromated Mo Kα (0.71073 Å) radiation and a low-temperature device. All calculations were performed using the SHELXTL (ver. 6.12) and SHELXL-97. The structures were solved by direct methods and successive interpretation of the difference Fourier map, followed by full-matrix least-squares refinement (against F2). Moreover, an empirical absorption correction using SADABS (ver. 2.03) was applied to all structures. All non-hydrogen atoms were refined anisotropically and the contribution of the hydrogen atoms, in their calculated positions, was included in the refinement using a riding model. The residual electron densities were located close to the heavy atom iridium in all structures, even when an absorption correction was applied. The thf molecules, (1) and (3), and the pentane molecule (2) in the crystal lattice are slightly disordered and a high number of restraints are used to achieve a satisfactory wR2 value. Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication No. CCDC-622198 (1), CCDC-622199 (2) and CCDC-622200 (3). Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: (+44) 1223-336-033; e-mail: deposit@ccdc.cam.ac.uk).
1 Selected bond lengths and angles for the bis(amido)iridate [Ir(trop2dach-2H)]− (average of two crystallographically independent molecules: Ir–N: 1.97025; Ir–ct 1.9985, CC 1.44275, ∑(N) = 353.4°, φ = 8.1°.
2 [Rh(I)(trop2dach)]+/[Rh(0)(trop2dach)]: E½1 = −1.83 V; [Rh(0)(trop2dach)]/[Rh(−I)(trop2diamine)]−: E½2 = −2.27 V.